Article Text

Abstract
Nucleoside reverse transcriptase inhibitors (NRTIs) remain the cornerstone of highly active antiretroviral therapy (HAART) combination regimens. However, it has been known for some time that these agents have the potential to cause varied side effects, many of which are thought to be due to their effects on mitochondria. Mitochondria, the key energy generating organelles in the cell, are unique in having their own DNA, a double stranded circular genome of about 16 000 bases. There is a separate enzyme present inside the cell that replicates mitochondrial DNA, polymerase gamma. NRTIs can affect the function of this enzyme and this may lead to depletion of mitochondrial DNA or qualitative changes. The study of inherited mitochondrial diseases has led to further understanding of the consequences of mutations or depletion in mitochondrial DNA. Key among these is the realisation that there may be substantial heteroplasmy among mitochondria within a given cell, and among cells in a particular tissue. The unpredictable nature of mitochondrial segregation during cellular replication makes it difficult to predict the likelihood of dysfunction in a given tissue. In addition, there is a threshold effect for the expression of mitochondrial dysfunction, both at the mitochondrial and cellular level. Various clinical and in vitro studies have suggested that NRTIs are associated with mitochondrial dysfunction in different tissues, although the weight of evidence is limited in many cases. The heterogeneity in the tissues affected by the different drugs raises interesting questions, and possible explanations include differential distribution or activation of these agents. This article reviews the major recognised toxicities associated with NRTI therapy and evidence for mitochondrial dysfunction in these complications. Data were identified through searching of online databases including Medline and Current Contents for relevant articles, along with abstracts and posters from recent conferences in the HIV and mitochondrial fields.
- mitochondrial toxicity
- HIV therapy
Statistics from Altmetric.com
Introduction
Current guidelines for the management of HIV infected individuals recommend the use of combinations including at least three agents—highly active antiretroviral therapy (HAART). This term was coined because of the magnitude of the effects seen in the early clinical studies of regimens combining the new HIV protease inhibitors (PIs) with the established nucleoside reverse transcriptase inhibitors (NRTIs). In addition to profound effects on viral load and CD4 cell count, these regimens were shown to significantly delay disease progression and death. Analyses of population data have also shown reductions in morbidity and mortality associated with increased use of HAART. The initial enthusiasm for PI regimens has waned somewhat with the realisation that these agents may be associated with long term toxicity in the form of a metabolic syndrome that has become known as HIV associated lipodystrophy.1 This, coupled with the development of cross resistance between currently available agents in the PI class, has led to a search for other regimens that may be employed either before or after PI therapy. Other triple regimens that have been studied have replaced the PI by either a non-nucleoside reverse transcriptase inhibitor (NNRTI) or another NRTI. These “PI sparing” regimens have shown comparable efficacy on surrogate markers of HIV disease in clinical trials. However, whatever drug is chosen as the third agent, HAART combinations invariably contain two NRTIs as the backbone of the regimen. While there are some toxicities common to all NRTIs, such as the rare syndrome of acute lactic acidosis, there are also a wide variety of tissue specific toxicities with each agent, the precise pathophysiology of which is frequently unclear. In vitro studies have shown that NRTIs may inhibit the replication of mitochondrial DNA2 and thus it is believed that some clinical toxicities of these agents may be the result of impaired mitochondrial function or replication, as reviewed recently by Brinkman and colleagues.3–5 This article will review the evidence for NRTI induced mitochondrial dysfunction in the context of our current understanding of mitochondrial biology and possible management strategies that may be employed to minimise the clinical effects of these toxicities in the HIV infected individual, with a particular focus on recently identified toxicity.
Mitochondrial biology
STRUCTURE
The identification of filaments (mito) and grains (chondria) under the light microscope by scientists in the 19th century provided early clues, but it was only the advent of electron microscopy that led to the widespread visualisation of mitochondria as the sausage-shaped organelles as shown in figure 1 and familiar to students of medicine and biology the world over.6 While this classic picture has been accepted for a number of years, recent studies have shown it to be an oversimplification. Rather than always being present as discrete organelles, mitochondria have also been shown to form a highly integrated network, and to undergo what appears to be a frequent process of fusion and fission. In addition, depletion in mitochondrial DNA has been shown to cause morphological changes in mitochondria from cultured human cells, and a high energy demand or oxidative stress will induce proliferation of the mitochondrial network to satisfy the cell's energy needs.

Mitochondria and the cell.
FUNCTION
The main function of mitochondria is to produce energy for the cell in the form of adenosine triphosphate (ATP), via the process of oxidative phosphorylation (fig 2). Acetyl-CoA is generated either via glycolysis in the cytosol or β oxidation of fatty acids in the mitochondria. The passage of acetyl-CoA through the tricarboxylic acid cycle generates NADH and FADH2, which are powerful reducing agents. Oxidative phosphorylation takes electrons from these reducing agents and passes them down the electron transport chain, eventually reducing oxygen to water. The transport of electrons down the different components of the electron transport chain also leads to the pumping of protons out of the mitochondria. This creates an electrochemical gradient leading to the return of protons into the mitochondria via specific channels. As protons pass through this channel, an integral component catalyses the synthesis of ATP. The ATP is then exchanged with ADP from the cytosol by a specific carrier, the ADP/ATP translocator. In addition, while considering mitochondrial function it is important to acknowledge that mitochondria are known to participate in other cellular processes, particularly apoptosis. Thus, it can be seen that mitochondria are not only essential for energy generation within the cell but also function as key regulators of cellular survival.

Energy generation in mitochondria. Mitochondria generate energy by oxidative phosphorylation. If this is impaired, energy may be generated by anaerobic metabolism where pyruvate is converted to lactate, possibly leading to lactic acidosis.
GENOME AND REPLICATION
Uniquely among organelles, it was recognised back in the 1960s that mitochondria have their own DNA distinct from that of the cell. Mitochondrial DNA is a circular, double stranded DNA molecule of about 16 000 bases, coding for 13 polypeptides, 22 transfer RNAs (tRNAs), and two ribosomal RNAs (rRNAs).7 The gene products of mitochondrial DNA are quite limited, and the bulk of the organelle is actually encoded for by nuclear DNA (nDNA). Considering mitochondrial genetics, there are certain features that are highly significant.8 Firstly, mitochondrial DNA is maternally inherited. Paternal mtDNA copy number in sperm cells is low in number by comparison with the large number of mtDNA molecules in the oocyte. In addition, it appears that although paternal mtDNA is transferred during fertilisation, it is lost early in embryogenesis. One of the most important observations of mitochondrial genetics is that different mtDNA variants may coexist in a single cell, the state of heteroplasmy. It has also been noted that there is a genetic bottleneck in mitochondrial DNA at some point between oogenesis and development of the embryo. This means that although there may be a large degree of heteroplasmy in the mother, the restriction and amplification that occurs during the bottleneck results in a very small number of mtDNA variants ultimately populating the embryo. Considering the effects of heteroplasmy at a cellular and tissue level, we encounter two further features of mitochondrial genetics, replicative segregation and the threshold effect. Replicative segregation refers to the distribution of heteroplasmic mitochondria during cell division (fig 3), although it is now known that mitochondria do have some interaction with cytoskeletal components and thus this process is unlikely to be completely random. The threshold effect can also be seen in figure 3, since it is only when the population of mitochondria with altered mitochondrial DNA exceeds a certain threshold (usually quite high, >80%) that the cell shows evidence of mitochondrial dysfunction.9

Although homoplasty is the normal state, different populations of mitochondria may coexist in a cell, the state of heteroplasmy. Since segregation of mitochondria during cellular replication is almost random, daughter cells may have different ratios of normal to dysfunctional mitochondria to the parent cell. If the population of mutant mitochondria reaches a certain threshold (70% in this example, but this can be as high as 95% in some tissues), then the cell exhibits dysfunction.9
MITOCHONDRIAL DNA POLYMERASE
There are at least nine polymerases involved in the replication and maintenance of cellular DNA; however, only one, DNA polymerase gamma, is responsible for mitochondrial DNA replication. Human mitochondrial DNA polymerase is a family A DNA polymerase and was cloned and characterised by Ropp and Copeland in 1996.10 It has been shown that polymerase gamma is expressed and translated in cells which have been depleted of mitochondrial DNA.11 The process of mitochondrial DNA replication has recently been reviewed12 and will not be described here. Polymerase gamma has to perform both replication and repair for mtDNA although for some time it was believed that repair activity was absent. However, polymerase gamma has been shown to participate in base excision repair, and other repair proteins have also been shown to be present in mitochondria; thus the once common view that mitochondria had little or no capacity for DNA repair should be reconsidered.13
Mitochondrial disease
Mitochondrial disease can result from mutations or rearrangements in both mitochondrial14 and nuclear DNA,15 and generally involves post-mitotic tissues. The clinical presentation includes organs such as the central and peripheral nervous system, the bone marrow, skeletal and cardiac muscle, the gastrointestinal tract, the kidneys, the pancreas, and the liver. An important feature that relates to the heteroplasmy and threshold effects discussed above is the heterogeneity of the clinical presentation of many of these inherited defects.16 This is thought to relate to the mutation load in particular tissues of different individuals. For example, Kearns-Sayre syndrome, an encephalomyopathy, and Pearson syndrome, a disease of the pancreas and bone marrow, both result from the same deletion of nearly 5000 base pairs in mitochondrial DNA. A further example is the mutation in a tRNA gene at position 3243, which when present at high levels is associated with the MELAS syndrome (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes), but with maternally inherited diabetes and deafness when present at low levels. The critical significance of the level of mutant mitochondrial DNA in determining the clinical presentation has naturally led to a focus on the patterns and maintenance of heteroplasmy. There has been some controversy over whether mutant or normal mitochondria have a selection advantage, since results using cybrid cells have not been consistent. A recent model suggests that although damaged mitochondria may replicate as quickly as normal mitochondria, they are degraded more slowly and thus persist in post-mitotic cells, but mitotically active tissues may be able to rejuvenate the population.
NRTIs and mitochondrial DNA
After HIV has entered the cell, it is required to integrate with the host cell genome. To do this, it needs to convert single stranded viral RNA into double stranded DNA and this task is performed by the enzyme reverse transcriptase. The NRTIs resemble the natural nucleosides but do not have a free 3` hydroxyl group, and thus once they are added to the growing DNA chain, termination occurs. Many NRTIs have been investigated for anti-HIV activity, and some knowledge of the structure-activity relation of these agents has been established. Since these drugs resemble natural nucleosides, the potential for inhibition of DNA polymerases exists, although there are sufficient differences between the enzymes to enable selective inhibition to occur.
IN VITRO STUDIES WITH NRTIS
Early studies of NRTIs demonstrated minimal effect on DNA polymerase alpha, the main enzyme responsible for nuclear DNA replication, but polymerase beta and gamma were affected to some degree.17 The clinical significance of the inhibition of DNA polymerase beta is unknown, this enzyme synthesises short sections of DNA as part of a group of enzymes involved in repair. Recently it has been shown that Tat, a gene product of HIV, induces the expression of DNA polymerase beta.18 The effects of NRTIs on polymerase gamma have been studied and it appears logical that inhibition of this enzyme and chain termination would lead to mitochondrial DNA depletion, which upon falling below the critical threshold would lead to insufficient energy generation and subsequent cellular dysfunction.2 Initial in vitro studies with NRTIs examined the toxicity of these agents in murine bone marrow progenitor cells, since the first NRTI licensed for the treatment of HIV, zidovudine, was associated with anaemia and neutropenia in the clinic. These studies showed that zidovudine exhibited toxicity in these models, appearing to confirm what was observed in patients.19 Later studies in neuronal cell models showed that the ddC, ddI, and d4T caused toxicity, whereas AZT and 3TC did not, again reflecting what was seen in clinical practice.20 The recognition that NRTIs may interfere with mitochondrial DNA synthesis led to many studies evaluating these effects in vitro, recently reviewed by Kakuda.21 These studies suggested a ranking of ddC ≥ ddI ≥ d4T > 3TC > ZDV > ABC for effects on mitochondrial polymerase gamma. Martin and colleagues examined both the inhibition of polymerase gamma and the inhibition of mitochondrial DNA synthesis, since some correlation was expected.22 Although a similar ranking of the NRTIs for effects on mitochondrial DNA synthesis was noted, there was no clear correlation with the potency of mitochondrial DNA inhibition. For example, one agent studied (935U83) was a highly potent inhibitor of polymerase gamma and yet showed no effect of mitochondrial DNA synthesis.22
Considering all in vitro experiments, it is important to note that these studies can only provide information on the effect of a drug in a particular cell type under given experimental conditions. Sensitivity to drugs varies between cell types and there are many other factors involved. One that has obvious implications if the mechanism of NRTI toxicity is indeed incorporation into mitochondrial DNA, is removal by exonuclease activity. Gray and colleagues demonstrated that lamivudine (3TC) is a substrate for the exonuclease activity of polymerase gamma, thus it can be excised if incorporated.23 Studies with other NRTIs have shown that they are substrates for cytosolic exonucleases, but the exonuclease activity may be inhibited by high levels of the NRTI monophosphate.24
ACTIVATION OF THE NRTIS
Another important factor to consider is the anabolism of the NRTIs, since these agents need to be phosphorylated three times before they can be added to the growing DNA chain by HIV reverse transcriptase or other polymerases. This process is known to vary with the activation state of the cell, with stavudine (d4T) and zidovudine (ZDV) being more active in activated cells and other NRTIs being more active in resting cells. In addition, intermediary anabolites may be implicated in toxicity of NRTIs, as has been shown for ZDV monophosphate in a study of CEM cells.25 Considering the mitochondrial effects of NRTIs, it is important to note that many cellular kinases exist in both mitochondrial and cytosolic forms. In addition to their subcellular localisation, these kinases frequently differ in their substrate specificity and regulation through the cell cycle. Early studies of zalcitabine (ddC) suggested that the drug was phosphorylated in the cytosol and then transported into the mitochondria. In the neuronal cell model referred to above, it was noted that ddC was only phosphorylated to the monophosphate in mitochondria, compared with the monophosphate, diphosphate, and triphosphate in the cytosol.20 Other investigators have reported similar results, and transport activity for both dCTP26 and ddCDP-choline27 has recently been described, suggesting that mitochondrial toxicity due to ddC is the result of transport of anabolites into the mitochondria, rather than phosphorylation therein.
Although less relevant for ddC, studies with other NRTIs demonstrate that mitochondrial phosphorylation can be a factor in determining drug toxicity. Fialuridine (FIAU) was investigated for the treatment of hepatitis B but unfortunately led to profound liver toxicity in most patients exposed to the drug for longer than 10 weeks.28 The liver toxicity was a consequence of severe mitochondrial dysfunction and in vitro experiments showed that FIAU was a much better substrate for mitochondrial thymidine kinase (TK-2) than the cytosolic form (TK-1), although the fact that FIAU is not a chain terminator like other NRTIs and may be incorporated also played a significant part.29 The NRTIs zidovudine and stavudine are also activated by thymidine kinase, and initial studies suggested that they could inhibit both TK-1 and TK-2, although d4T was not shown to act as substrate for either of these enzymes.30 Subsequently it has been recognised that d4T is relatively poorly phosphorylated compared with thymidine and ZDV,31 and later studies have shown that d4T is indeed a substrate for these enzymes.32 In the neuronal cell model referred to above, d4T was shown to be phosphorylated to the triphosphate within the mitochondria.20 Considering other nucleosides, while the formation of ddA-monophosphate from didanosine (ddI) occurs through a slightly different enzymatic mechanism than other NRTIs, the second phosphorylation step is catalysed by adenylate kinase. This enzyme also exists as cytosolic (AK-1) and mitochondrial forms (AK-2), and thus the potential for differential inhibition exists. Our understanding of kinases involved in the activation of guanosine analogues, such as abacavir and DAPD, is not as far advanced in terms of a link between activation and toxicity, although studies with nucleosides used in cancer therapy have shown a direct relation between mitochondrial deoxyguanosine kinase activity and cytotoxicity.33
NRTI TRANSPORT
The entry of NRTIs into cells has been observed to occur at different rates, and there are many transport systems available for nucleosides. Since the phosphorylation of NRTIs may differ between subcellular compartments, it follows that movement of the drugs and their anabolites between the cytosolic and mitochondrial compartments is of considerable interest.34 Early studies with lamivudine (3TC) showed synergistic or additive activity against HIV in vitro, and also protection against the delayed mitochondrial toxicity associated with d4T, ZDV, ddC, and ddI.35 The protection conferred by 3TC in this study was thought to be due to interference with the uptake of the other agents into mitochondria. Subsequent experiments have shown that other “unnatural” NRTIs in the same class, such as L(−)Fd4C, also show similar properties.36 Considering NRTI transport into mitochondria, the studies discussed above with 3TC and L(−)Fd4C suggest that this process may be inhibited, although whether NRTI anabolites can be exported from mitochondria remains unclear. Further understanding of the activation and transport of the NRTIs within different subcellular compartments may lead to molecules or strategies in which efficacy can be enhanced and toxicity reduced.
HIV AND MITOCHONDRIA
It is known that many of the toxicities associated with NRTI therapy may also be related to HIV infection itself, but it is not often appreciated that there is also known to be a direct interaction between HIV and mitochondria. Early studies showed that HIV RNA could be found in mitochondria of infected cells, and that there were mitochondrial alterations in patients with the acute HIV syndrome and stable infection. More recent work has shown that the HIV TAT protein may promote mitochondrially induced apoptosis,37 consistent with our knowledge of the importance of this process in the immune cell destruction caused by the virus. A specific interaction between the HIV viral protein R and the mitochondrial permeability transition pore complex (PTPC) has recently been demonstrated by Jacotot and colleagues,38 and with the recognised involvement of the PTPC in apoptosis it seems likely that HIV affects the immune system at least in part by interacting with mitochondria leading to programmed cell death. Such viral effects are not uncommon; indeed the hepatitis B virus protein X has also been shown to interact with a component of the PTPC.
Clinical toxicity of the NRTIs
NRTIs are associated with a wide spectrum of toxicities, many also caused or exacerbated by HIV itself. Since the tissue involvement and clinical presentation often resembles aspects of inherited mitochondrial disease, and it is known that NRTIs may affect mitochondrial function, many authors have proposed that mitochondrial toxicity of the NRTIs is the underlying pathophysiology behind most of these toxicities.3 Lewis and Dalakas put forward the “polymerase gamma hypothesis” in their review,39 suggesting that the manifestations of NRTI toxicity relate to the combined effects of four principal factors. Firstly, the tissue must have some dependence on oxidative phosphorylation; secondly, the NRTI must pass into the tissue itself; thirdly, the NRTI must be phosphorylated by cellular kinases; and, finally, it must inhibit polymerase gamma activity by competing with the natural substrate or by chain termination.39 The data reviewed earlier identify some refinements that may be made to this hypothesis such as the role of phosphorylation in different subcellular compartments, particularly the mitochondrion itself. More fundamental is the lack of correlation between polymerase gamma inhibition and mitochondrial DNA depletion as identified by Martin and colleagues.22 In addition, the neuronal cell model showed that the neurotoxic effect of d4T did not correlate with mitochondrial DNA depletion, whereas there was a correlation between the effect of ddC and mitochondrial DNA levels.20 A full reappraisal of the polymerase gamma hypothesis is beyond the scope of this review, although our extended understanding of the many processes involved in NRTI toxicity warrants some expansion of the initial ideas. The major toxicities associated with NRTI therapy will now be reviewed and evidence for a mitochondrial pathophysiology discussed.
HAEMATOLOGICAL TOXICITY
HIV infection and AIDS are known to be associated with significant haematological toxicity, including anaemia, neutropenia, and thrombocytopenia.40 In addition, studies with zidovudine have shown that this drug may compound the haematological toxicity of HIV and lead to the independent development of anaemia and neutropenia.41 Consistent with these observations, the incidence of anaemia or neutropenia in mildly or asymptomatic adults treated with zidovudine was between 1.1% and 9.7%, whereas in adults with AIDS or AIDS related complex it ranged from 15% to as high as 61%.41 This toxicity is generally reversible and may be managed by dose reduction or drug withdrawal. Other interventions also appear to confer varying degrees of benefit, including growth factors for neutropenia,42 and recombinant haemoglobin43 or erythropoietin44 for anaemia. In vitro studies also confirm the haematological toxicity of HIV and zidovudine,19 although the mechanism of this toxicity is unclear. One study suggested that permeation of the drugs into canine bone marrow progenitor cells might be an indicator of drug specific toxicity, since although ZDV permeation was itself slow, ddI was even slower and ddC did not permeate at all.45 Törnevik and colleagues suggested that the cytotoxic effect correlates with ZDV monophosphate levels,25 Faraj and colleagues confirmed these findings and did not demonstrate a correlation between toxicity and mitochondrial DNA inhibition,46 suggesting that this effect may occur through some other mechanism. Although it is known that mitochondrial dysfunction may result in haematological toxicity, it seems possible that zidovudine associated haematological toxicity may result from alternative effects, perhaps on haem metabolism or gene expression. Both zidovudine and stavudine treatment are known to be associated with macrocytosis, although the mechanism and clinical significance is unclear.47 HIV infection is also known to be associated with thrombocytopenia,48 possibly through the action of specific viral strains in the bone marrow, and this has been shown to improve with zidovudine treatment.49
MYOPATHY
HIV infection may be associated with myopathy at all stages of the disease.50 It was also noted that zidovudine therapy may be associated with myopathy in the early studies with this agent,51 although distinguishing the disease and drug related myopathies has proved difficult.52 Simpson and colleagues analysed data from a placebo controlled study of zidovudine,53 and conducted a prospective myopathy substudy in a large trial of combination therapy.54 In the first study, ACTG 016, five out of 279 (1.8%) zidovudine treated patients had a composite myopathy diagnosis, compared with none in the placebo group. ACTG 016 examined a dose of 200 mg zidovudine every 4 hours (1200 mg/day), and three instances of dose reduction in the five patients with a composite myopathy diagnosis led to improvements in creatine kinase levels, but no improvement in strength. However, CK levels were also observed to improve independent of dose reduction, and in these instances improvement in strength was noted on one occasion.53 The second study, ACTG 175, was a placebo controlled comparison of ZDV or ddI monotherapy and ZDV/ddC or ZDV/ddI dual therapy in 2467 patients with HIV infection. A myopathy substudy was also conducted in the 1067 antiretroviral naive participants in this trial. No significant differences were observed between the treatment arms for adverse events related to myopathy, although the ZDV/ddC group had a reduced incidence of myalgia compared with the other groups. Although site diagnoses in this study were made without predefined criteria, it is noteworthy that only six cases of myopathy were identified, four on ddI monotherapy, one each on ZDV/ddI and ZDV/ddC dual therapy, and none on ZDV monotherapy.54 The lack of an association between ZDV therapy and myopathy in this study may in part relate to the dose administered, since the dose of ZDV used in this study was half that in ACTG 016 (600 mg/day v 1200 mg/day).
The data reviewed above illustrate that drug related myopathy is quite rare, particularly with currently used doses, and a number of studies have attempted to explain the pathophysiology of this disorder. Benbrik and colleagues studied the effects of ddI, ddC, and ZDV on cultured human muscle cells and showed that although ZDV was the most potent inhibitor of cell proliferation, ddC and ddI were the most potent inhibitors of mitochondrial function.55 Depletion of mitochondrial DNA has been reported in patients with zidovudine related myopathy, and this has been shown to be reversible on drug withdrawal.56 The histopathological assessment of myopathy has proved somewhat controversial, particularly with regard to the presence of “red ragged fibres” and abnormalities in mitochondrial morphology. Since there is frequently little correlation between these observations and the presence of myopathy, their use in isolation is not recommended.57 Chariot and colleagues have shown that the histological assessment of zidovudine myopathy may be improved by looking cytochrome c oxidase activity,58 and studies in rats have reported similar observations. Many inherited mitochondrial diseases are known to be associated with myopathy,59 and the changes observed in mitochondrial DNA levels in clinical and laboratory studies of zidovudine myopathy strongly support a mitochondrial pathophysiology for this effect, although it should be acknowledged that there are data suggesting other mechanisms.60 Finally, it is worth noting that HIV infection may rarely be associated with rhabdomyolysis, and with the increasing use of cholesterol lowering drugs in patients with HIV infection, the potential myotoxicity of the HMG CoA reductase inhibitors should be noted since cases of rhabdomyolysis associated with these agents in HIV patients have been reported.61
CARDIOMYOPATHY
Abnormal cardiac pathology was identified in postmortem studies of AIDS patients in the mid 1980s, and subsequent studies have shown that cardiac involvement in HIV infection is by no means uncommon.62 The spectrum of cardiac disease in HIV infection has been reviewed elsewhere and will not be considered further here.63 It is known that mitochondrial dysfunction may frequently be associated with heart disease.64 In vitro studies have identified that cardiac mitochondrial DNA polymerase may be inhibited by zidovudine,65 and studies in rats have shown that the drug may also induce ultrastructural changes in cardiac myocytes.66 A recent study by Lewis and colleagues demonstrated that zidovudine and HIV infection led to the independent development of cardiomyopathic changes in a transgenic mouse model,67 although the dose of zidovudine used in this study was significantly higher than that used clinically in HIV infected patients (∼200 mg/kg v 8 mg/kg). Lipshultz and colleagues studied cardiac structure and function in HIV infected children and found no association between any changes and treatment with zidovudine.68 In a more recent study, the same group studied infants exposed to zidovudine perinatally and again noted an absence of cardiac dysfunction.69 Others have reported an association between cardiomyopathy and therapy with ZDV, ddC or ddI,70 but the major part appears to be played by HIV disease or other as yet unidentified factors.71
NEUROPATHY
Peripheral neuropathy has long been recognised as a complication of HIV infection, the incidence of which increases with the degree of immunosuppression.72 Three of the currently licensed NRTIs, ddC, ddI and d4T, have also been associated with the development of distal symmetrical polyneuropathy in clinical studies.73 It is difficult to distinguish HIV and drug related neuropathy, although drug related neuropathy is more likely to be painful, have an abrupt onset, and rapid progression.74 The diagnosis of neuropathy should follow a comprehensive neurological history and examination, including assessment of other risk factors such as concomitant neurotoxic medication or nutritional deficiency.73 In the neuromuscular substudy of ACTG 175 discussed above, only half of the site diagnoses were confirmed as drug related by a review using defined criteria.54 The incidence of drug related neuropathy was significantly greater in the ZDV/ddC arm, but not different in the ddI, ZDV, or ZDV/ddI arms.54 The duration of treatment with the neurotoxic NRTIs increases the likelihood of the development of drug related neuropathy, with 25% of ddC treated patients developing this complication after >9 months of therapy.74 Studies with ddI have shown that this agent is associated with the development of neuropathy but at lower frequency than ddC or d4T, and the reported rates have been similar to non-neurotoxic agents in some studies.74 In the large parallel track study of d4T, neuropathy was reported in 17% of patients receiving d4T 40 mg/day compared with 23% of patients receiving 80 mg/day.74 Although there is limited information on the risk associated with combinations of neurotoxic NRTIs, Moore and colleagues have recently reported an analysis in 1116 patients on the Johns Hopkins HIV database. This study showed that the risk of peripheral neuropathy in patients treated with ddI and d4T was 3.5-fold greater than for patients treated with ddI alone in a multivariate model adjusted for other factors.75 Hydroxyurea was shown to increase the risk of peripheral neuropathy still further, since patients taking ddI/d4T hydroxyurea were shown to have a 7.8-fold greater risk than ddI alone,75 findings that have also been confirmed by other groups.76 Since studies have shown that the development of neuropathy with ddC, ddI, and d4T is dose related, management by dose reduction or discontinuation is normally recommended.74 In addition to treatment with analgesics, tricyclic antidepressants, and topical anaesthetics specific interventions have been examined. Although recent studies of recombinant human nerve growth factor77 and vibratory stimuli78 have not shown significant efficacy, a pilot study of lamotrigine reported some improvements in symptoms.79 Famularo and colleagues noted that 12 HIV infected patients with peripheral neuropathy on regimens including ddC, ddI, or d4T had acetyl carnitine deficiency compared with controls with no disease or non-drug related neuropathies.80 Subsequent to this, Hart and colleagues recently reported a small, uncontrolled investigation into the use of l-acetyl-carnitine (1500 mg twice daily) for the treatment of drug related neuropathy and showed improvements in symptoms and evidence of peripheral nerve regeneration in the four patients studied.81 Carnitine and its derivatives are important intermediaries in the transport and utilisation of fatty acids by mitochondria. More recently, Cergnul and colleagues reported a small, short term, placebo controlled study of topical aspirin in diethyl ether, during which the patients treated with this solution reported improvements in pain relief.82
In vitro studies have confirmed the neurotoxic potential of ddC and ddI, and correlations with mitochondrial DNA depletion were observed.20 However, although d4T exhibited neurotoxicity in the same study, there was no correlation with mitochondrial DNA depletion. A study in rabbits showed mitochondrial alterations in peripheral nerves after ddC treatment,83 but similar studies with ddI and d4T showed no neurotoxic effect in the same species,84 although ddI has been shown to cause neurotoxicity in rats given very high doses. The well described neuropathies associated with inherited mitochondrial diseases85 and the evidence discussed above do suggest a role for mitochondrial dysfunction in the pathogenesis of drug related neuropathy but inconsistencies do exist. The benefit of l-acetyl-carnitine supplementation also supports this mechanism but it is clear that larger, well controlled studies are needed before it can be widely recommended, and other therapies for the management of neuropathy are urgently required.
PANCREATITIS
Individuals with HIV infection are at greater risk for the development of pancreatitis as a result of immunodeficiency and exposure to a variety of pancreatotoxic agents.86 These include drugs used to treat opportunistic infections, such as pentamidine, and drugs to treat the HIV infection itself, principally didanosine. The reported incidence of pancreatitis with didanosine varies. Maxson and colleagues suggested it was relatively common with clinical pancreatitis present in 23.5% of their patients and asymptomatic elevations of amylase and lipase in an additional 39.2%.87 However, it should be noted that most of the patients in this study were taking both pentamidine and didanosine, thus additive or synergistic toxicity would be predicted to account for the higher rates observed.87 Other studies suggest an incidence ranging from 4%–7% at currently recommended doses, with rates increasing at higher doses of ddI.86 Fatal cases of pancreatitis have been reported,88 and mortality ranges from 6% at current doses to 17% at higher doses. The ACTG 5025 study examined intensification of d4T/ddI/IDV with hydroxyurea and reported that two of the patients in the hydroxyurea arm developed fatal pancreatitis, two other cases in the non-hydroxyurea arm survived.89 More recently, Moore and colleagues reported a fourfold increased risk for pancreatitis when hydroxyurea was used with ddI or ddI and d4T. They also reported a similar incidence associated with either ddI or d4T alone and an increased risk of pancreatitis if these two agents were used together.90 Asymptomatic elevations in serum pancreatic enzymes occur more frequently than clinical pancreatitis, and these should be monitored, particularly if other risk factors are present. If pancreatitis has developed in the past, permanent discontinuation of ddI is suggested since rechallenge often results in recurrent disease.86 There are limited data suggesting an association with other NRTIs, the incidence with ddC therapy appears to be <1%,91 and although early paediatric studies described cases in children receiving 3TC, many of these children had a previous history of pancreatitis or were receiving concurrent pancreatotoxic medication.86 A recent comparative study of ZDV/3TC and ddI monotherapy in 471 paediatric HIV patients reported no cases of pancreatitis in the ZDV/3TC arm and four in the ddI arm,92 and pancreatitis has rarely been reported in adults treated with 3TC.93 Cases of pancreatitis associated with severe lactic acidosis have been reported,94 and mitochondrial dysfunction is known to be associated with pancreatic abnormalities,95 but the evidence supporting a mitochondrial pathophysiology for ddI associated pancreatitis is limited at present. Early studies in a rat model of pancreatitis demonstrated no changes even at extremely high doses of ddI,96 although a more recent study of a pancreatic cell line suggested a dose dependent mitochondrial toxicity of ddI in this system.97 Since it has been observed that the protease inhibitors may lead to significant increases in plasma triglyceride levels and it is also known that raised triglyceride levels are a risk factor for pancreatitis, these parameters should be monitored since cases of pancreatitis have been reported under these circumstances.98
LACTIC ACIDOSIS
Severe liver toxicity, manifesting as acute lactic acidosis with evidence of hepatic steatosis is probably the most worrisome toxicity of the NRTIs, since it can often be fatal and progression may be rapid. It was first identified in the era of NRTI monotherapy but cases have continued to be reported with dual and triple combination therapy. Brinkman and colleagues have recently reviewed this area4 and other reviews have been published previously99 so this section will concentrate on recent data.
When a cell is unable to generate enough energy through oxidative phosphorylation, anaerobic respiration occurs via the conversion of pyruvate to lactate in the cytoplasm. This also results in excess production of hydrogen ions, which if uncontrolled, may lead to a cellular and subsequently metabolic acidosis. Clearance is normally performed by the liver and kidneys but if the production is excessive or these organs are damaged, accumulation of lactate and hydrogen ions may occur and severe lactic acidosis may result. Lactic acidosis is a known feature of many mitochondrial disorders9 and steatosis results from inhibition of fatty acid oxidation, leading to the accumulation of lipid droplets. In vitro studies and recent reports showing evidence of mitochondrial dysfunction after detailed clinical and laboratory investigations in patients with lactic acidosis associated with d4T and ddI100 or ZDV101 confirm a mitochondrial pathophysiology. Cases of severe lactic acidosis have been reported with all NRTIs, and the incidence of the acute syndrome is fortunately quite rare, around 1.3 per 1000 person years, although it should be noted that the initial data from which this rate was calculated were probably inadequate.4
Recently a number of groups have studied the prevalence of symptomatic hyperlactataemia and others have surveyed large cohorts for the prevalence of lactate levels above the upper limit of normal. Before reviewing these data, it is worth mentioning that lactate measurement is fraught with problems and care needs to be taken over sampling and handling to avoid misleading values.102 In an extended follow up of their earlier paper,103 Lonergan and colleagues have described 33 patients with decompensated or symptomatic hyperlactataemia associated with NRTI treatment in a large cohort.104 The clinical presentation and lactate elevations were milder than in other studies of acute lactic acidosis. Symptoms included abdominal pain, nausea, and distension, and only one patient required hospitalisation. Antiretroviral therapy was discontinued in these patients until lactate levels normalised (median time 49 days).The incidence of this syndrome was observed to be about 13-fold greater than that quoted for acute lactic acidosis, and it was also noted to be particularly associated with d4T containing regimens, with 31/33 cases on d4T containing therapies. Seventeen patients have restarted therapy, 16 replacing d4T with ABC (n=10), ZDV (n=4), or both (n=2). All patients taking 3TC restarted this drug in their new regimen, and there have been no recurrences of the syndrome to date.104 Gerard and colleagues described 14 cases of decompensated hyperlactataemia in a cohort of 871 patients; the incidence was 0.8% per year which rose to 1.2% per year if only patients on d4T containing regimens were included.105 The clinical presentation was similar to that described by Lonergan and colleagues, muscle biopsies from four of five patients showed ultrastructural and biochemical evidence of mitochondrial dysfunction. These data suggest that patients may experience a milder form of decompensated lactic acidosis more frequently than the acute syndrome described previously. Hence, signs and symptoms should be monitored since this syndrome may also be associated with significant morbidity. Regarding the association with d4T containing regimens, it is important to interpret such data with caution since there may be confounding biases which have not been identified in such studies and the number of cases are too small to draw definitive conclusions. Many other large studies have also investigated the prevalence of hyperlactataemia; however, it is important to distinguish these studies from those of Lonergan and Gerard, since these larger studies have reported any patients with raised lactate levels, not just those with symptoms. Across these studies, there is a higher incidence of hyperlactataemia on d4T containing therapies (table 1).106–111 In addition, some studies primarily focused on lipodystrophy have also noted an association between d4T therapy and hyperlactataemia.112 Saint Marc and colleagues noted that patients switched from d4T to either ZDV or ABC had significant reductions in their plasma lactate levels, which were elevated on d4T therapy.113, 114
Cohort studies of raised lactate levels or anion gap
While it remains clear that acute lactic acidosis, decompensated hyperlactataemia, and compensated hyperlactataemia may be associated with all NRTIs the data reviewed above suggest a greater risk associated with d4T therapy. Prospective studies of prevalence and resolution will be required to confirm these observations, although the evidence from multivariate analyses of large cohorts appears consistent. The OZCOMBO I study, a randomised comparison of ZDV/3TC, d4T/3TC, and d4T/ddI with indinavir described significantly increased lactate levels with d4T/ddI arm compared with the other two arms, although it should be noted that the level of follow up in this analysis was poor.115 A key question that remains to be answered is whether the presence of asymptomatic hyperlactataemia is an indicator of a predisposition to develop symptomatic lactic acidosis, or whether it reflects the current level of interest in this parameter and thus more frequent monitoring and reporting. The comparative rarity of decompensated lactic acidosis compared with the high frequency of patients with lactate elevation but no symptoms suggests that routine lactate monitoring is unlikely to be helpful in this regard. A high degree of awareness of related symptoms would appear to be the best approach at present. Treatment of lactic acidosis has been reviewed by Brinkman,4 and generally involves the administration of what might be termed mitochondrial supportive therapy such as thiamine, riboflavin, ubiquinone, and acetyl-carnitine in addition to cessation of antiretroviral therapy and intensive support. Doses and efficacy are unclear and since it is likely that cessation of NRTI therapy is the most significant factor contributing to observed improvements controlled studies are required. Brinkman and colleagues have suggested a protocol in the absence of standardised recommendations.116 Dichloroacetate (DCA) has been studied for the treatment of lactic acidosis and was found to have no benefit it terms of decreased mortality in one large study.117 However, through its action on pyruvate dehydrogenase, DCA directly reduces lactate production and more recent case reports have suggested that this agent may be useful in NRTI associated lactic acidosis.118
LIPODYSTROPHY
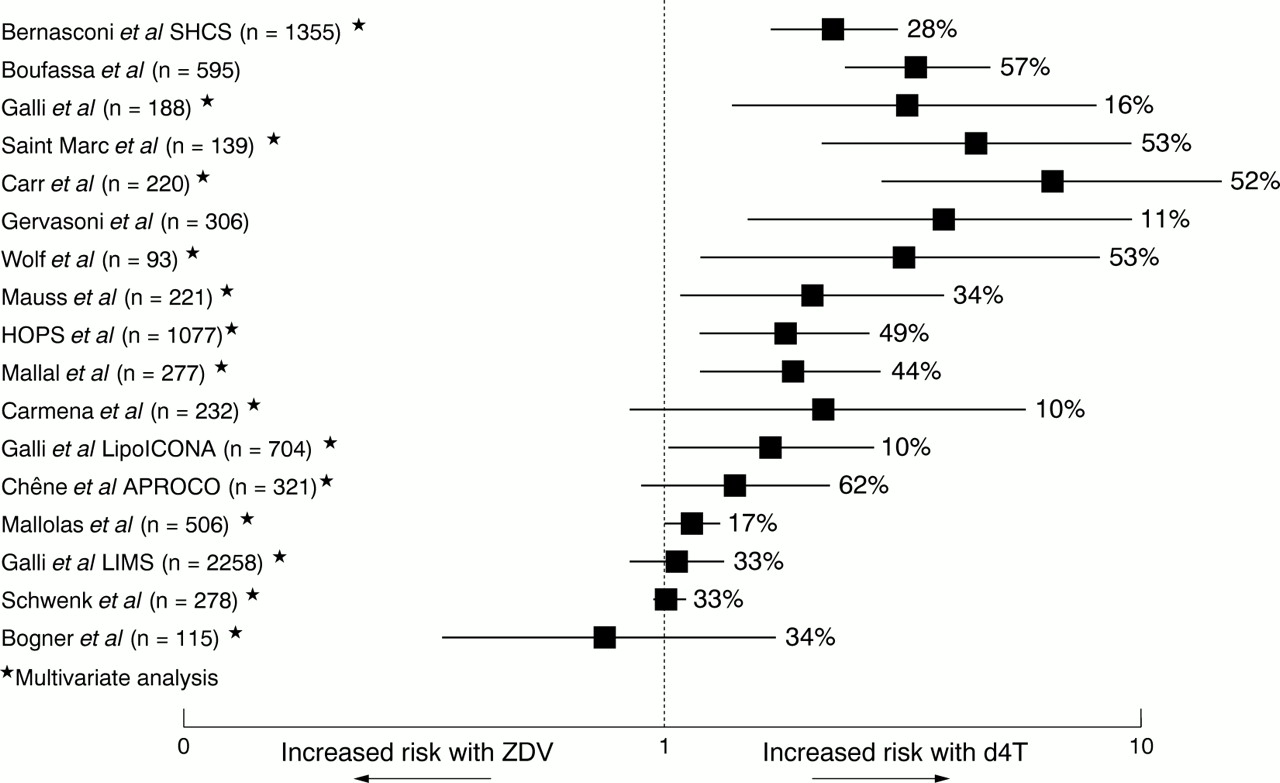
Reports of metabolic and body habitus changes in HIV patients led to the description of what has become known as HIV associated lipodystrophy. Initial studies associated this syndrome with the protease inhibitors although subsequently it has become clear that the NRTIs, particularly d4T, also play a part. A detailed discussion of lipodystrophy is beyond the scope of this article; recent reviews of lipodystrophy119 and HIV associated lipodystrophy1 have been published. This section will describe the studies associating NRTI therapy with lipodystrophy, since Brinkman and colleagues120 and Kakuda and colleagues121 have proposed that this may be related to mitochondrial toxicity. Before reviewing the data it is worth noting that a consensus definition for lipodystrophy is still under development and there are many other factors that may influence the development of metabolic or body habitus changes in an individual. Therefore, it must be remembered that lipodystrophy is undoubtedly a syndrome with a multifactorial aetiology. Table 2 presents the findings from both cohort studies and randomised, controlled trials that have examined the association of NRTIs with lipodystrophy.115, 122–140, 167 It can be seen that overall duration of NRTI therapy and d4T therapy in particular are associated with the development of lipodystrophy. Since time on therapy has been identified as a risk factor the association with d4T could simply be because it was the most recently used nucleoside. However, most of the studies conducted rigorous multivariate analyses to control for confounding factors such as time on therapy, and the independent effect of d4T remained significant. Furthermore, the evidence from cohort studies (fig 4) is consistent with that from randomised controlled trials in therapy naive patients—for example, the ALBI study. In this study, d4T/ddI was associated with a significantly greater risk of lipodystrophy than ZDV/3TC in both analyses, whether of all patients as randomised at the start of the study (54% v 20%, p=0.001), or those patients who remained on NRTIs alone (52% v 10%, p=0.02).138 Some authors have examined switching patients from d4T to other NRTIs to improve lipodystrophy. In an extended follow up of an earlier study,113 Saint-Marc et al reported 59 patients with subcutaneous fat wasting on d4T therapy (18 on NRTI therapy and 41 on PI therapy), where d4T was replaced by either ZDV or ABC. This was shown to result in major improvements in peripheral fat wasting in most patients independent of concomitant PI use (83% in the NRTI group and 71% in the PI group) after 12 months. Fat wasting was assessed by objective measurement (abdominal and mid-thigh CT scan) and reductions in serum lactate levels were also observed.114 Polo and colleagues also observed improvements in fat wasting and metabolic parameters in 10 patients switched from d4T/ddI to ZDV/3TC while maintaining indinavir.141 A more recent report described improvements in facial fat wasting in 11 patients who discontinued d4T, nine switched to ZDV or ABC.142 The data reviewed above strongly support a significant role of d4T among the NRTIs in the development of subcutaneous fat wasting, and provide encouraging early evidence with regard to reversibility on switching to other NRTIs; however, the results of ongoing randomised studies are awaited to confirm these findings.
Cohort studies and randomised controlled trials of lipodystrophy with NRTIs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cohort studies have shown that d4T (stavudine) is associated with a greater risk of lipodystrophy, primarily subcutaneous fat wasting, compared with ZDV (zidovudine). This figure shows the relative risk for D4T versus ZDV from studies where this information was given or could be calculated. It should be noted that the multivariate analyses denoted by an asterisk are the most reliable indicators, since they have controlled for possible confounding variables such as previous therapy duration Note: % refers to LD prevalence.
Brinkman and colleagues proposed that NRTI associated lipodystrophy may be related to mitochondrial toxicity partly due to the similarities between Madelung's disease and some of the clinical features of the lipodystrophy syndrome.120 While Saint-Marc and colleagues noted a reduction in plasma lactate levels as lipodystrophy improved114 and Carr and colleagues described a syndrome of lactic acidaemia and peripheral fat wasting,112 both of these observations also related to the presence or absence of d4T therapy, which is known to be associated with a higher risk for the development of both hyperlactataemia and peripheral fat wasting. More recently, Walker et al143 and Shikuma et al144 have demonstrated that fat biopsies from patients with lipodystrophy show depletion of mtDNA compared with controls, and Mallal and colleagues have demonstrated ultrastructural abnormalities of mitochondria in similar tissue.145 The mean loss of mtDNA reported by Walker and colleagues was 44%, below what might normally be considered a threshold for expression of dysfunction. However, it is important to recognise that the threshold effect applies to individual cells within a tissue; since a biopsy is likely to include both normal and abnormal cells (fig 3) this lower average level of depletion supports rather than discounts some mitochondrial effect. Paulik and colleagues have described an association between metabolic complications associated with NRTIs and changes in the expression of genes and markers of oxidative stress in animal models and cell culture.146 They also reported that antioxidants such as ascorbate and tocopherol ameliorated these adverse effects, suggesting future studies of these agents may be warranted.
RENAL TOXICITY
Adefovir, a nucleotide analogue, was shown to induce nephrotoxicity in a significant proportion of patients after longer than 6 months' exposure.147 Recent experiments have demonstrated that adefovir is a substrate for the human renal organic anion transporter 1, and is thus concentrated in the cells of the proximal tubule.148 This has been shown to result in increased cytotoxicity in vitro, although the mechanism of this cytotoxicity is unclear.148 The clinical presentation of renal involvement in inherited mitochondrial disease is similar to that noted for adefovir toxicity,149 and adefovir is known to inhibit mitochondrial DNA polymerase,150 but it is currently unknown whether the nephrotoxicity of adefovir is mitochondrial in nature. Adefovir is no longer being pursued for the treatment of HIV, although lower doses are being studied for the treatment of hepatitis B infection. No particular association with other nucleoside analogues has been reported, although a case of nephrotoxicity with ddI was reported some years ago151 and one with d4T/3TC more recently,152 although the latter may have been associated with decompensated hyperlactataemia.
NRTIS AND PERINATAL TRANSMISSION
Zidovudine therapy has been widely used for the prevention of mother to child transmission and significant reductions have been observed in the number of new infections as a result of this and other interventions. Blanche and colleagues recently reported eight cases of mitochondrial dysfunction in uninfected children exposed to zidovudine and lamivudine in a clinical study for the prevention of perinatal transmission.153 The evidence for mitochondrial dysfunction in these children was not comprehensive, only two had evidence of histological or histochemical features of mitochondrial disease and mitochondrial DNA levels were normal in the three children for whom data were available. In a more recent report, the same authors have described 16 cases strongly suspected of mitochondrial dysfunction.154 Since the initial reports, there have been extensive efforts among other groups to look for similar cases, however various analyses have failed to identify any deaths similar to those reported by Blanche and colleagues.155–158 Nevertheless, this study raised important questions around the risk-benefit ratio of the prevention of perinatal transmission using antiretrovirals. Further research and follow up must continue, particularly considering the use of other NRTIs for this indication and to inform clinical management, since elevated lactate levels in infants are common and may be associated with a variety of causes but could also indicate mitochondrial dysfunction in the presence of other findings. Taking these data in context, the clear public health benefits associated with reductions in perinatal transmission have meant current recommendations for antiretroviral use remain unchanged, although continued vigilance is warranted.159
Management of mitochondrial toxicity
Since NRTI associated mitochondrial toxicity is acquired during NRTI treatment, cessation of the offending agent or dose reduction may be sufficient to limit the toxicity as has been discussed when these clinical toxicities have been reviewed above. There are a variety of strategies that may be employed to treat mitochondrial disease,160 but they are generally poorly studied and are frequently used on the basis of a theoretical rationale alone. Brinkman has reviewed the evidence supporting the use of supplements such as thiamine, riboflavin, ubiquinone, and acetyl-carnitine,4 and it would certainly appear reasonable to employ these agents in patients with severe mitochondrial disease, such as lactic acidosis. However, whether they might be used concomitantly with antiretroviral therapy to delay the onset of mitochondrial toxicity, if that is indeed the pathophysiological mechanism for the toxicity concerned, remains to be answered by prospective studies.
Mitochondrial toxicity of new agents
With the inexorable development of resistance to all agents used to treat HIV, new agents are continually being investigated. With the known potential for mitochondrial toxicity with NRTIs, detailed studies are required although the inconsistencies discussed previously with various in vitro and animal models make comprehensive evaluation difficult. Fialuridine (FIAU) was an investigational nucleoside analogue for the treatment of hepatitis B, and although initially well tolerated, after 12–14 weeks many patients experienced severe multiorgan mitochondrial toxicity and died, despite receiving emergency liver transplants.28 Studies in dogs and monkeys had failed to predict this toxicity, although later studies in woodchucks confirmed the profound mitochondrial toxicity of this agent.161 The withdrawal of adefovir discussed earlier and the recent suspension in the development of F-ddA illustrate the difficulties in developing new NRTIs for the treatment of HIV. Didanosine (ddI) is actually converted to ddA as it is anabolised, and lodenosine (F-ddA) is a fluorinated version of this molecule with better bioavailability and enhanced potency against HIV in vitro. In vitro studies did not identify appreciable effects on mitochondrial DNA levels or lactate production,162 but a phase II study of F-ddA in combination with d4T and indinavir was stopped when one patient died and others showed evidence of liver or kidney problems.163 As has been discussed earlier, the use of hydroxyurea to boost the efficacy of NRTIs has been shown to be associated with an increased risk of peripheral neuropathy and pancreatitis.75, 76, 89 Since hydroxyurea leads to a reduction in the intracellular levels of natural nucleosides, NRTIs are more likely to be incorporated by reverse transcriptase, thus their efficacy may be enhanced. However, the clinical evidence suggests that hydroxyurea also increases the likelihood of NRTI incorporation into mitochondrial DNA, thus the possible increase in efficacy must be balanced with an increased risk of toxicity.
Considering licensed agents, the most recent nucleoside analogue to be approved for the treatment of HIV infection, abacavir, showed minimal potential for mitochondrial toxicity in laboratory studies.164 This has subsequently been confirmed in clinical studies of over 25 000 patients which have yet to identify an association between the drug and significant long term toxicity.165, 166 However, abacavir is associated with a hypersensitivity reaction in approximately 4% of patients which may be life threatening on rechallenge.166 The improvements in lipodystrophy and lactate levels described previously when patients were switched from d4T to either ZDV or ABC suggest the drug may have utility in such circumstances.
Summary
Nucleoside reverse transcriptase inhibitors (NRTIs) remain the cornerstone of highly active antiretroviral therapy (HAART) combination regimens. However, it has been known for some time that these agents have the potential to cause varied side effects, many of which are thought to be due to their effects on mitochondria.
Mitochondrial toxicity does appear to be the related to many of the adverse effects associated with treatment with these agents. However, we should be careful not to assume that all NRTI toxicities are due to this particular mechanism. In considering the evidence reviewed previously, while lactic acidosis and myopathy seem clearly mitochondrial in nature, we start to encounter difficulties in associating neuropathy with this pathophysiology owing to the different responses of the neurotoxic agents in various studies. Anaemia and neutropenia seem more likely to be a direct effect of zidovudine or one of its anabolites, and we only have one in vitro study to suggest ddI associated pancreatitis might be mitochondrial in nature. Perhaps unsurprisingly, although lipodystrophy is clearly associated with NRTIs and particularly stavudine, we are still in the process of describing the many different pathways that may be involved hence it is premature to assign one particular mechanism.
Considering management of toxicity, while dose reduction or discontinuation of the causative agent may lead to improvements or resolution of the side effects associated with that particular drug, this may not always be an option. Supplements such as acetyl-carnitine, riboflavin, thiamine, and coenzyme-Q all have a theoretical rationale for use in these situations and have been shown in studies to have some benefit, although their prospective use is not yet justified.
Continued research into the pathogenesis of NRTI associated toxicity, coupled with the significant advances being made in mitochondrial research, should lead to improved understanding which will facilitate interventions to manage these toxicities, and aid the development of newer agents for which these toxicities are absent or minimised.
Acknowledgments
I would like to thank David Nolan, Simon Mallal, and Dominic Paes for their valuable comments during the preparation of this manuscript.
Conflict of interest: AJW is employed by GlaxoSmithKline Research and Development, part of GlaxoSmithKline Plc, who manufacture and market many currently licensed treatments for HIV infection.