Article Text

Statistics from Altmetric.com
Case presentation (Dr B Peters)
A 40 year old heterosexual black African man presented to the accident and emergency department with a 4 minute episode of loss of consciousness with associated incontinence. On inquiry, he gave a 1 month's history of left sided headache, weight loss, fever, and a right axillary swelling. He had a medical history of recurrent malaria, typhoid as a child, drainage of groin abscess in 1999, and severe acne. An HIV antibody test had been negative in 1993. On admission he was taking ciprofloxacin 500 mg twice daily, prescribed by a primary care physician as treatment for the axillary infection; he had recently completed a course of isotretinoin as treatment for acne. He worked as a delivery driver, drank no alcohol, and was a non-smoker. He had been resident in the United Kingdom for 7 years.
On examination he was pyrexial, temperature 38.5°C, had oral candida, severe acne, and a 4 cm diameter infected sebaceous cyst in the right axilla. There was no lymphadenopathy and examination of the respiratory, cardiovascular, abdominal, and nervous systems was normal.
Investigations on admission to hospital included a computed tomograph (CT) scan of the head (with contrast) which showed a single ring enhancing lesion in the left basal ganglia with surrounding oedema and mid-line shift. A diagnosis of cerebral toxoplasmosis was made. Subsequently, an FDG PET (positron emission tomography) scan and cranial magnetic resonance image (MRI) supported this diagnosis. Other investigations showed HIV-1 antibodies detected, cytomegalovirus IgG positive, DAT negative, Toxoplasma gondii IgG > 1:1024, serum cryptococcal latex agglutination (CRAG) negative, CD+ T lymphocyte count 4 cells ×106/l, HIV viral load 190 000 copies/ml, hepatitis A IgM and IgG negative, hepatitis B surface antigen negative, hepatitis C IgG negative. Urea and electrolytes were normal as was a full blood count. Liver function tests showed normal bilirubin and alkaline phosphatase, the AST was 66 (normal range = 0–55) IU/l and γGT = 137 (normal range = 0–72) IU/l.
What would your initial management plan be?
Discussant (Dr R Miller)
I would begin empirical therapy for cerebral toxoplasmosis, based on the findings of cranial imaging and the high titre of anti-toxoplasma antibodies. Although in many case series, single lesions are more likely to be due to lymphoma,1 here you have the additional evidence from FDG PET scanning to support the diagnosis of toxoplasmosis. The assessment of response to empirical therapy would be based on clinical defervescence, neurological improvement, and reduction in size of the lesion on repeat cranial imaging. The treatment regimen of first choice would be sulphadiazine with pyrimethamine and folinic acid. I would avoid using dexamethasone, unless the patient was obtunded or had signs of raised intracranial pressure (bradycardia, widened pulse pressure, Cheyne-Stokes respiration), as clinical/imaging improvement may occur from steroid induced reduction in oedema because of either toxoplasmosis or another cause such as tuberculosis or lymphoma. This improvement may be misleading and result in delayed diagnosis of the correct disease process. Anti-epilepsy medication should be given perhaps with sodium valproate rather than phenytoin, in order to avoid a potential drug-drug interaction with antiretroviral therapy.2
Once I had microbiological identification of the cause of the axillary abscess, I would modify antibiotic therapy for this. I would treat the oral candidiasis with amphotericin lozenges and I would screen the patient to exclude tuberculosis as an intercurrent diagnosis. In this particular situation, I would be tempted to delay starting antiretroviral therapy until the patient had stabilised. This would help avoid diagnostic confusion which might arise from side effects of the antiretrovirals as well as avoiding the possibility of an immune reconstitution syndrome The timing of the introduction of antiretrovirals is uncertain, and a case could be made for introducing them earlier.
Case presentation (Dr Peters)
We also thought the most likely diagnosis was cerebral toxoplasmosis. Treatment for this was with sulphadiazine/pyrimethamine and folinic acid; dexamethasone was also given because of concerns about the mid-line shift on cranial imaging. Phenytoin was given to prevent further fits. Sodium fusidate and clarithromycin were given for the axillary abscess and amphotericin lozenges for candida. Fourteen days after admission antiretroviral therapy was started with stavudine and ritonavir/saquinavir. What do you think about this regimen?
Discussion (Dr Miller)
This combination cannot really be considered ideal as first line therapy. There is some evidence for the efficacy of dual protease inhibitor/single NRTI (nucleoside reverse transcriptase inhibitor) containing regimens
Case presentation (Dr Peters)
The day after starting antiretroviral therapy liver function tests were noted to be abnormal; alkaline phosphatase = 123 IU/l, ALT = 326 IU/l, γGT = 1069 IU/l. A clotting screen was normal and an ultrasound scan of the abdomen revealed a normal sized liver with a homogeneous, slightly increased echogenicity, consistent with fatty changes. No intrahepatic bile duct dilation was seen and the gall bladder and common bile duct had normal appearances.
In light of this information, what would your management be now?
Discussion (Dr Miller)
The abnormal liver function tests and ultrasound appearances are surprising and suggest there may be another (missed) diagnosis. The time course for the liver function test deterioration is far too soon for this to be due to antiretroviral therapy or to an immune reconstitution syndrome. I think I would want to consider the possibility of a viral hepatotrophic infection. We know the patient is hepatitis B sAg negative, but is he core Ab positive? Is there a risk of reactivation? I would also want to check hepatitis A and C as well as cytomegalovirus (CMV), EBV (Epstein-Barr virus), and herpes simplex virus (HSV) serology. Other infectious aetiologies need consideration, given his ethnicity and lifetime travel history. The lack of organomegaly and peripheral blood abnormalities does not exclude tuberculosis or a disseminated fungal infection such as histoplasmosis. Lymphoproliferative disease including lymphoma, should be considered and even possibly a late presentation of a hereditary liver condition such as α1 antitrypsin deficiency or Kinnear-Wilson syndrome. Finally, I would be concerned about a potential hepatotoxic effect of medication: isotretinoin is associated with biochemical abnormalities of liver function in 15% of patients, and sulphadiazine may produce abnormal liver function tests as a result of drug induced granulomatous inflammation. The liver ultrasound rules out hepatitis as this is associated with normal or reduced echogenicity, no focal lesions are seen, excluding tumour and abscess and there is no perihepatitis. Hyperechogenicity may be found in patients with diabetes mellitus or long standing hyperlipidaemia and may also be found coincidentally in some patients of African origin so the finding is non-specific. In this patient, I wonder if sulphadiazine is the cause of the liver function abnormalities. I would consider stopping this drug and substituting clindamycin.
Case presentation (Dr Peters)
We thought this to be most likely the result of drug toxicity and so stopped sulphadiazine and commenced clindamycin. Six weeks later, his liver function test had normalised, apart from the γGT which was 357 IU/l. At this time, the viral load remained high at 119 489 copies/ml and the CD4+ T lymphocyte count was 52 cells ×106/l. Adherence issues were discussed with the patient at length. By September 2000, the viral load was 2363 copies/ml and CD4+ T lymphocyte count was 139 cells ×106/l. A resistance test showed no genotypic resistance mutations and antiretroviral therapy was intensified with addition of abacavir in December 2000. By March 2001 the liver function tests had deteriorated, ALT = 511 IU/l and γGT = 853 IU/l; bilirubin, alkaline phosphatase, and prothrombin remained normal. Repeat hepatitis B serology and hepatitis C polymerase chain reaction (PCR) were negative. A liver ultrasound scan showed a fatty liver with no dilatation of the biliary tree. At this point, a test was performed which suggested a diagnosis. What was this test?
Discussion (Dr Miller)
I might consider performing a liver biopsy as this might reveal the precise cause of the liver function abnormalities—for example, a disseminated infection as I mentioned earlier. Against performing this investigation is the fact that the procedure has a higher morbidity, and indeed, mortality in HIV infected patients compared to the general population.3 Considering all the information you have given me so far, the concept of drug induced mitochondrial toxicity would have to be considered. One of the most simple and cost effective tests would be to send a blood sample for measurement of urea and electrolytes. By doing this the anion gap [(serum sodium + serum potassium) − (serum bicarbonate + serum chloride)] can be calculated. If the gap was increased, it would guide you towards checking a resting, uncuffed venous lactate. An elevated serum lactate, typically ≥5 mmol/l with an increased anion gap, suggests a metabolic acidosis due to lactic acidosis, which should be confirmed with an arterial blood gas sample—to obtain the pH and standard bicarbonate (both are low in a metabolic acidosis). An elevated serum lactate with a normal anion gap indicates hyperlactataemia, not lactic acidosis. Once other causes of a raised lactate such as sepsis or a myocardial infarction have been excluded, this finding would be strongly suggestive of mitochondrial toxicity.
The investigation most likely to give a diagnosis in my view is a liver biopsy. A less invasive investigation, which might suggest the diagnosis, is an uncuffed lactate. I suppose I am here to have my head “put on the block,” so I will opt for a venous lactate measurement.
Case presentation (Dr Peters)
The test was an uncuffed venous lactate. The result = 3.7 mmol/l (normal range = 0.7–2.2 mmol/l). The test was repeated a week later and lactate had increased to 5.3 mmol/l. The diagnosis at this stage was either hyperlactataemia secondary to antiretroviral drug induced mitochondrial dysfunction affecting the liver or hepatic steatosis induced by antiretroviral therapy. A liver biopsy was performed; what do you think it showed?
Discussion (Dr Miller)
Let me begin by saying what it does not show! I do not think it will show granulomata or cholestasis and there won't be any evidence of focal or diffuse tumour infiltration. I think special stains will be negative confirming there is no infection causing this presentation. Finally, I am sure there will be no α1 antitrypsin globules and no deposits of copper associated protein. I think, really sticking my neck out, the liver biopsy is going to show macrovesicular and microvesicular steatosis, with mild portal inflammation. I am led to this by the liver ultrasound scan appearances, the lack of clinical progression with time, had the diagnosis been an infection or tumour, and the temporal association with antiretroviral therapy. If my diagnosis is correct, then I would stop the NRTI drugs stavudine and abacavir.
Pathology (Professor S Lucas)
The liver biopsy showed macrovesicular and microvesicular steatosis with many areas of focal necrosis (fig 1). There was no evidence of lymphoproliferative disease and special stains were negative for fungi and mycobacteria. This pattern of liver damage is very consistent with drug induced steatosis. Although electron microscopy was not performed in this case, in clinically similar cases we have observed abnormally shaped mitochondria. This conforms with the hypothesis of drug induced mitochondrial toxicity.
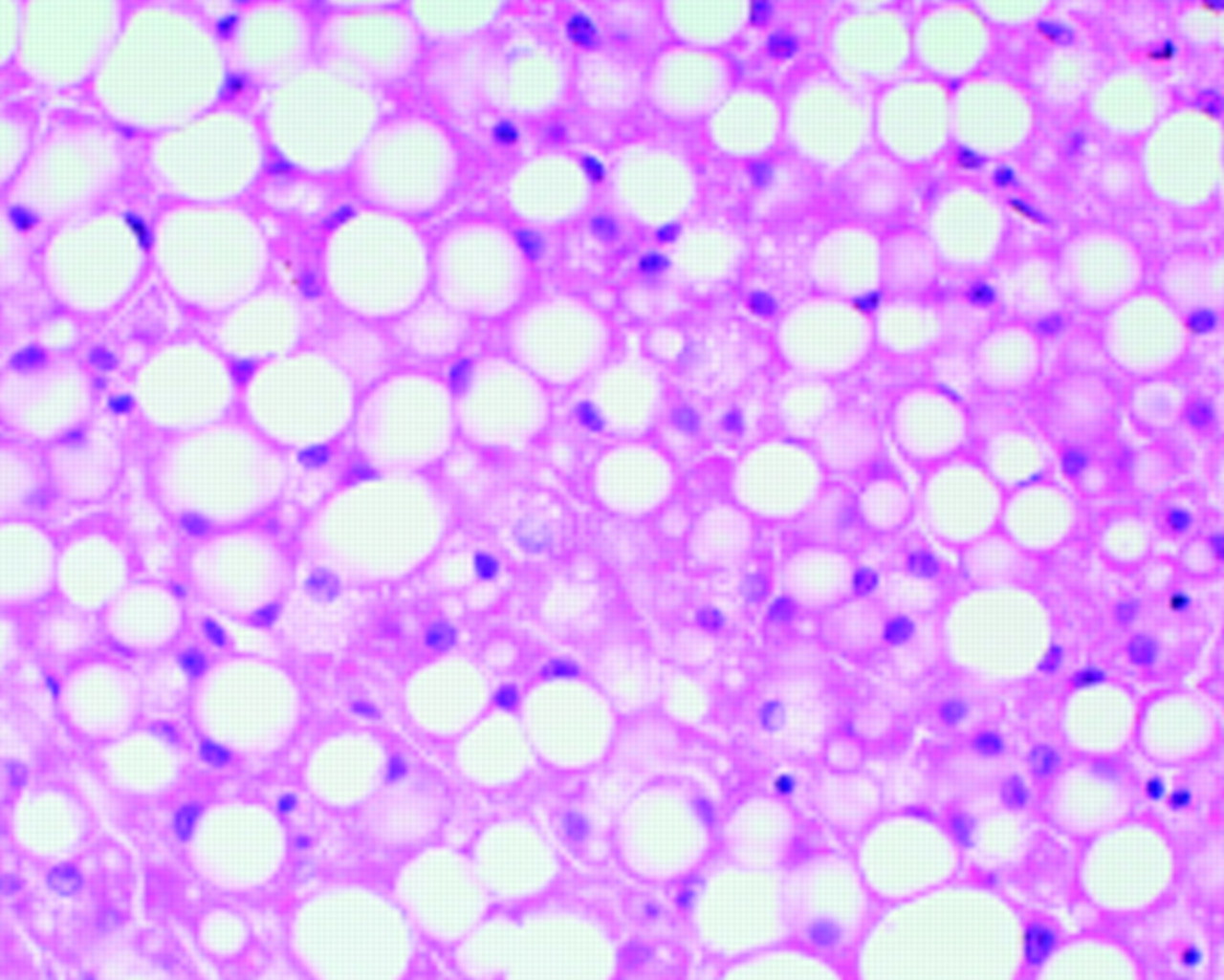
{kind=link}
Liver showing predominantly macrovesicular steatosis. Haematoxylin and eosin; magnification ×400.
Case presentation and discussion (Dr Peters)
The stavudine and abacavir were discontinued and the venous lactate returned to normal over the next 14 days. The clinical diagnosis is hepatic mitochondrial toxicity secondary to NRTI therapy.
The link between NRTI therapy and mitochondrial damage was first suggested in 1990, in relation to myopathy induced by zidovudine.4–6 Subsequently, pancreatitis and peripheral neuropathy were reported in patients receiving didanosine; peripheral neuropathy has also been described with zalcitabine and stavudine.7 These presentations are also thought to be manifestations of NRTI induced mitochondrial toxicity.7 The first fatal case of lactic acidosis induced by an NRTI was described in 1993.8 This presentation—hepatic steatosis and lactic acidosis—appears to be uncommon. In one retrospective cohort study, the incidence was 1.3 cases per 1000 person years.9 In contrast, hyperlactataemia (which is usually asymptomatic) appears to be much more common—in one study it was estimated to be 20.9 cases per 1000 person years.10 In a cross sectional study of 211 asymptomatic HIV infected patients, 39 (13.2%) had mild hyperlactataemia (lactate = 2.1 − 5.0 mmol/l).11 Of these patients, 35 (89%) were receiving NRTI therapy. It is important to have clear definitions. Brinkman has suggested the following: mild hyperlactataemia (lactate = 2.1 − 5.0 mmol/l), serious hyperlactataemia (lactate >5 mmol/l), and lactic acidosis (lactate >5 mmol/l and bicarbonate <20 mmol/l).11
Discussion (Dr Miller)
There is no practical screening test for this condition, as mild hyperlactataemia in an asymptomatic patient does not predict for the development of lactic acidosis.7, 11 Other tests such as measurement of the lactate:pyruvate ratio are impractical for screening. The clinician needs a high index of suspicion to make the diagnosis. If any HIV infected patient receiving HAART, which contains an NRTI develops non-specific symptoms—for example, malaise, tiredness, nausea and vomiting, then a resting uncuffed lactate should be measured, as well as the urea and electrolytes. If the lactate is elevated, the anion gap is widened and the bicarbonate is low then an arterial blood gas sample should be taken to confirm lactic acidosis.
In a patient with mild hyperlactataemia and no or only minor symptoms, no specific intervention is necessary. Close monitoring in the clinic is indicated and, in some cases, the elevated lactate normalises spontaneously11; in others, there is no deterioration in lactate levels. With severe hyperlactataemia, most patients are symptomatic. Management here would be to withdraw antiretroviral therapy and the lactate level frequently returns to normal, although in some patients this can take many weeks.10 This suggests that there is not only an abnormality of lactate generation but also an abnormality of clearance by the liver, in the context of NRTI induced hepatic toxicity. Patients with lactic acidosis are usually markedly symptomatic. This presentation has a high mortality, of the order of 80%, and intensive medical support is required. The mainstay of these interventions is bicarbonate infusions and haemofiltration; occasionally mechanical ventilatory support will be needed. Several adjunctive therapies have been used. The rationale for use of these agents is based on knowledge of the enzymes and biochemical pathways involved in the oxidative mitochondrial pathway and extrapolation of clinical experience in patients with inherited mitochondrial diseases.12 There are anecdotal case reports and small case series which support the use of l-carnitine, ubiquinone (coenzyme Q), and riboflavin (vitamin B-2) as adjunctive therapy.12
Summary (Dr Peters)
There has been a paradigm shift in HIV management. In the early years of the HIV pandemic we were managing the complications of HIV induced immunosuppression, for example, Pneumocystis carinii pneumonia. Now, with effective antiretroviral regimens we are managing the complications of treatment of HIV itself. The weapon we use against HIV can sometimes turn in on us. Clinicians should have a high awareness of drug induced toxicities, such as hyperlactataemia, and be prepared to investigate and manage this condition.
Key points
-
Routine measurement of venous lactate in asymptomatic HIV positive patients is of no practical value.
-
An elevated venous lactate (2.1–5.0 mmol/l) in an asymptomatic patient does not predict for development of lactic acidosis.
-
In patients with severe hyperlactataemia (lactate >5.0 mmol/l) despite discontinuation of NRTI drugs, the venous lactate may take several weeks to return to normal.
-
Antiretroviral therapy should be discontinued immediately in patients presenting with lactic acidosis. Intensive medical support is indicated and adjunctive therapy may be tried.
Acknowledgments
Dr D Pao and Dr C Watson abstracted the patient's clinical record and prepared the case for presentation.
References
Footnotes
-
Source of funding: Nil.
-
Conflict of interest: Nil.