Article Text

Abstract
Objectives Drug-resistant Neisseria gonorrhoeae are now a global public health threat. Direct transmission of antibiotic-resistant gonococci between individuals has been proposed as a driver for the increased transmission of resistance, but direct evidence of such transmission is limited. Whole-genome sequencing (WGS) has superior resolution to investigate outbreaks and disease transmission compared with traditional molecular typing methods such as multilocus sequence typing (MLST) and N. gonorrhoeae multiantigen sequence (NG-MAST). We therefore aimed to systematically investigate the transmission of N. gonorrhoeae between men in sexual partnerships using WGS to compare isolates and their resistance to antibiotics at a genome level.
Methods 458 couples from a large prospective cohort of men who have sex with men (MSM) tested for gonorrhoea together between 2005 and 2014 were included, and WGS was conducted on all isolates from couples where both men were culture-positive for N. gonorrhoeae. Resistance-determining sequences were identified from genome assemblies, and comparison of isolates between and within individuals was performed by pairwise single nucleotide polymorphism and pangenome comparisons, and in silico predictions of NG-MAST and MLST.
Results For 33 of 34 (97%; 95% CI 85% to 100%) couples where both partners were positive for gonorrhoea, the resistance-determining genes and mutations were identical in isolates from each partner (94 isolates in total). Resistance determinants in isolates from 23 of 23 (100%; 95% CI 86% to 100%) men with multisite infections were also identical within an individual. These partner and within-host isolates were indistinguishable by NG-MAST, MLST and whole genomic comparisons.
Conclusions These data support the transmission of antibiotic-resistant strains between sexual partners as a key driver of resistance rates in gonorrhoea among MSM. This improved understanding of the transmission dynamics of N. gonorrhoeae between sexual partners will inform treatment and prevention guidelines.
- gonorrhoea
- antibiotic resistance
- modes of transmission
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Men who have sex with men with gonorrhoea are usually infected with the same strain of Neisseria gonorrhoeae as their partner, including those with multisite infection.
Partner isolates have identical antibiotic resistance determinants, and thus when a man is diagnosed with antibiotic-resistant gonorrhoea, his partner should also be treated for resistant gonorrhoea.
Whole-genome sequencing is a highly discriminant technology that can be used to identify gonorrhoea transmission networks.
Introduction
Increasing antimicrobial resistance in Neisseria gonorrhoeae is a major public health problem globally, with few effective therapeutic options remaining.1 While men who have sex with men (MSM) have been repeatedly linked to transmission of resistant N. gonorrhoeae,2 the drivers of antimicrobial resistance have not been completely defined. Molecular epidemiological approaches to the study of N. gonorrhoeae are critical to understanding the spread of resistance. Previous studies have used a variety of molecular typing methods, including N. gonorrhoeae multiantigen sequence typing (NG-MAST),3 multilocus sequence typing (MLST),4 and more recently whole-genome sequencing (WGS).5–8 These studies indicated relationships exist between some clonal types, such as NG-MAST 1407, and antibiotic resistance in N. gonorrhoeae at a population level, suggesting dissemination of resistance mechanisms through sexual networks is driving antibiotic resistance. However, there has been limited examination of the genomic epidemiology of defined transmission events to confirm this. Although it is assumed that transmission of these resistance elements occurs between partners, it has not been investigated at a person-to-person level, with the alternate hypothesis being that resistance arises de novo in an individual after acquiring gonorrhoea, with subsequent treatment selecting out drug-resistant strains.
Most resistance in N. gonorrhoeae results from the acquisition of point mutations in key genes encoding antibiotic targets.1 For example, resistance to penicillins and cephalosporins can develop through mutations in penA, the gene encoding penicillin-binding protein 2, with resistant isolates harbouring mosaic alleles with numerous amino acid alterations generally acquired through homologous recombination. Similarly, mutations in other genes including mtrR and the mtrR promoter region, penB, pilQ and ponA can also confer resistance to penicillins. Fluoroquinolone resistance is predominantly conferred by mutations in gyrA and parC, encoding subunits of the key quinolone targets, DNA gyrase and DNA topoisomerase IV, respectively.9 Macrolide resistance is more complex, with some mutations in the 23S rRNA target resulting in low-level resistance (eg, C2611T) and others in high-level resistance (eg, A2059G), with the number of mutated alleles of the four copies of 23S in N. gonorrhoeae also influencing the degree of resistance.10 Acquired resistance such as erm genes encoding rRNA methylases and tetM-carrying or blaTEM-carrying plasmids also circulate among gonorrhoea isolates, while overexpression of efflux pumps can also contribute to resistance.1
To understand the development and acquisition of antimicrobial resistance at the individual and sexual partner levels, we collected detailed epidemiological data and investigated gonorrhoea isolates from a series of male sexual partners using WGS. We compared the genomes of isolates cultured from men within sexual partnerships and analysed isolates from different sites within individual men, to understand the genomic diversity present within individuals and partnerships and to identify transmission of the genetic determinants of antimicrobial resistance at an individual level.
Methods
Patient selection
Male sexual partners included in this study were those who attended the Melbourne Sexual Health Centre, Australia, together on the same day between September 2005 and September 2014 and where both men in the partnership had N. gonorrhoeae cultured from at least one anatomical site: urethral, pharyngeal and/or rectal, irrespective of symptoms. Every individual attending the clinic was required to complete questions on their recent sexual history using a computer-assisted self-interview at registration. This included questions regarding the number and sex of recent sexual partners and condom use in the last 3 months, and sex overseas in the preceding 12 months. Individuals were also asked whether their sexual partner was attending the clinic on the same day, allowing male couples to be identified.
Specimen collection
Screening for gonorrhoea was undertaken using pharyngeal and rectal swabs for asymptomatic MSM, in addition to a urethral swab for MSM with urethritis. N. gonorrhoeae colonies were selected from the primary culture plate for speciation, antimicrobial susceptibility testing and subsequent DNA extraction. Antimicrobial susceptibility testing was performed using agar plate dilution as previously described for penicillin, ceftriaxone, ciprofloxacin, tetracycline, spectinomycin and azithromycin, using break points determined by the Australian Gonococcal Surveillance Programme (online supplementary table S1).11 12
Supplementary file 1
Whole-genome sequencing
WGS was performed on extracted DNA using Illumina libraries and protocols (Illumina, San Diego, California, USA). Raw sequence data have been uploaded to the Sequence Read Archive under the study accession PRJEB17738. In brief, sequencing data for each isolate were analysed and compared using read alignment to identify single nucleotide polymorphisms (SNPs), and de novo draft genome assembly for pangenome and gene allele comparisons. Comparisons of isolates were conducted using in silico MLST, in silico NG-MAST and a maximum likelihood phylogeny inferred from core genome SNPs after removing recombination. Previously described genetic determinants of antimicrobial resistance were identified and compared between isolates.1 13 Bioinformatics methods are detailed in the online supplementary appendix.
Statistics
Descriptive statistics of patient characteristics and N. gonorrhoeae isolates were reported. The 95% CIs were calculated by using exact binomial distribution in Stata 13.1.
Results
Characteristics of the study population
In total, there were 458 male couples where both men within the partnership were tested for N. gonorrhoeae infection by culture from at least one site. Excluding recurrent presentations, 34 couples (68 men) were identified where both partners had N. gonorrhoeae cultured from at least one site. Thirty-one (91%) of the 34 couples had at least one partner with urethral symptoms and N. gonorrhoeae cultured from the urethra. WGS was conducted on 94 isolates cultured from clinical samples. Table 1 shows clinical and microbiological characteristics of the individuals and isolates included in the analysis. A full list of isolates is provided in online supplementary table S2.
Characteristics of MSM within partnerships and their Neisseria gonorrhoeae isolates
Genomic mediators of resistance
All but two (98%) isolates were resistant to penicillin by agar dilution, with eight penicillin-resistant isolates showing phenotypic presence of a beta-lactamase (penicillinase-producing N. gonorrhoeae). Six (7%) isolates showed decreased susceptibility to ceftriaxone, 41 (45%) were resistant to ciprofloxacin, while 7 (8%) were tetracycline-resistant. No resistance to azithromycin or spectinomycin was detected.
Penicillin-binding protein mutations were the most prevalent resistance mechanisms, although a diverse range of mutations were seen. Seventy-nine isolates had non-mosaic penA alleles, which were associated with susceptibility to ceftriaxone. Ten isolates had identical mosaic penA XXXIV alleles and were all NG-MAST types 1407 or 4822, although this mosaic allele was only associated with decreased susceptibility to ceftriaxone for 6 of the 10 isolates. Three other isolates had interspecies mosaic penA XLI alleles likely to be derived from N. meningitidis.8 Characteristics of these 13 isolates with mosaic penA are shown in table 2. In addition to penA mutations, several isolates had mutations in mtrR (adenine deletion from 13 bp inverted repeat in the promoter region, and G45D substitution in mtrR) and ponA (L421P), which may also have contributed to penicillin resistance. The eight phenotypically identified penicillinase-producing isolates were found to carry bla TEM-1. Several mutations in the quinolone resistance-determining regions of gyrA (S91F, D95G) and parC (D86N, S87R, S87N, E91K, E91G) were identified, mediating ciprofloxacin resistance. All tetracycline-resistant isolates were found to carry tetM genes.
Resistance mechanisms in isolates with mosaic penA alleles
MSM are infected with the same strain as their partner
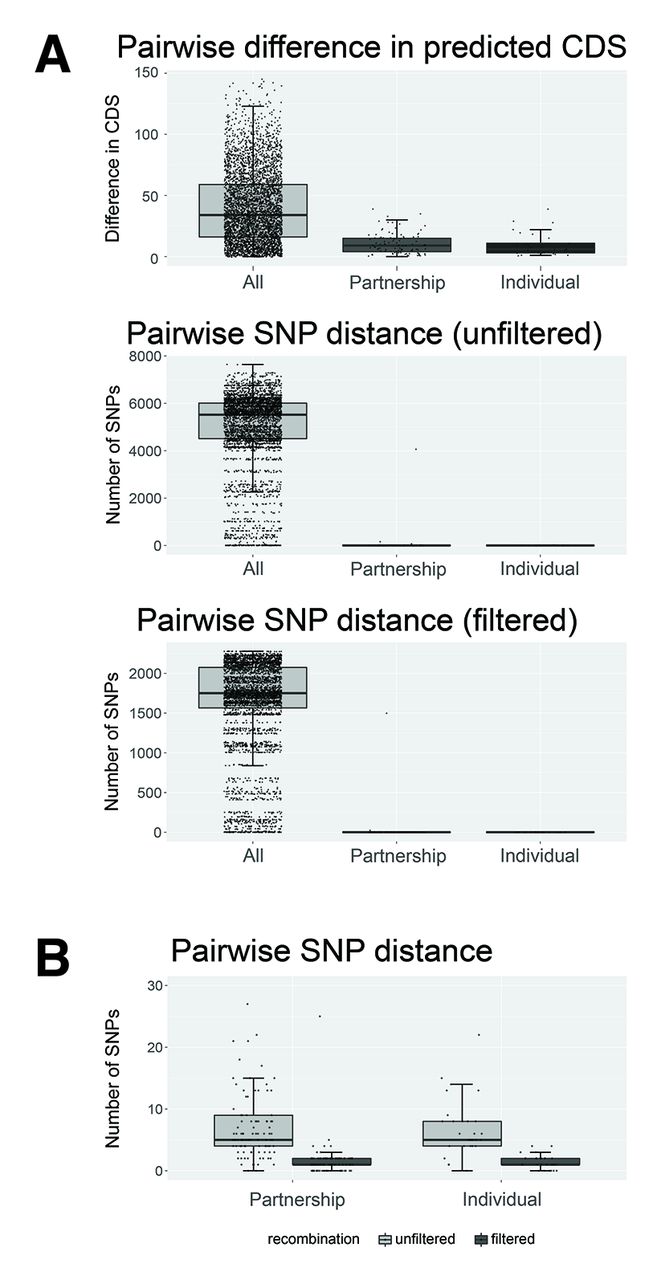
For 33/34 (97%; 95% CI 85% to 100%) couples, isolates from men in a sexual partnership were identical by NG-MAST and MLST. Each man’s isolates were also indistinguishable from his partner’s isolates using WGS, with the diversity between partner isolates being the same as the diversity of N. gonorrhoeae within an individual. Phylogenetically, the isolates from men in a given partnership were more closely related to each other, compared with isolates from men outside of the partnership (figure 1). The same was true when partner isolates were compared based on gene content. The difference in number of predicted coding DNA sequence (CDS) regions between isolates from partners (median=9, IQR 4–15) was lower than the difference in CDS regions between any two randomly selected isolates from the broader study population (median=34, IQR 16–59) (figure 2). The number of SNPs was also considerably lower between male partners (unfiltered median=5, IQR 4–9; filtered for recombination median=1, IQR 1–2) than with other men (unfiltered median=5521, IQR 4510–6011; filtered for recombination median=1751, IQR 1565–2075).
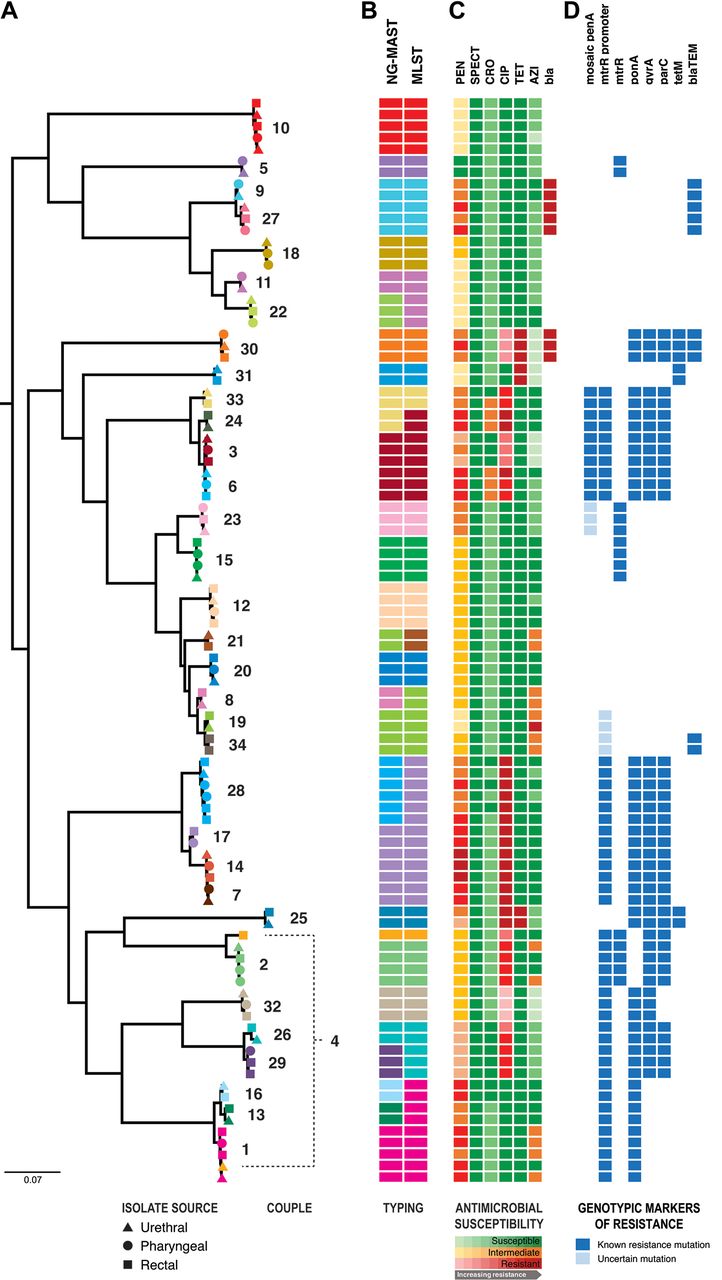
Phylogenetic, molecular sequence typing and antimicrobial susceptibility/resistance comparisons of 94 gonococcal isolates from 34 male couples. From left to right: (A) maximum likelihood phylogenetic tree of the Neisseria gonorrhoeae sequences after filtering for recombination. All major nodes had >70% bootstrap support. Isolates derived from the same couple have been coloured the same to illustrate isolates within partnerships, with each couple numbered. Shapes represent infection site: urethral (triangle), pharyngeal (circle) and rectal (square) isolates. (B) The tree is shown adjacent to molecular typing data from in silico sequence analysis for the two-gene multiantigen sequence typing (NG-MAST) and the seven-gene multilocus sequence typing (MLST) schemes. The same NG-MAST or MLST types are represented by the same colours. (C) Phenotypic susceptibility profiles are shown as a heatmap within susceptible (green), intermediate (orange) and resistant (red) categories, where progressively darker colours represent higher minimum inhibitory concentrations within the categories. AZI, azithromycin; bla, phenotypic beta -lactamase production; CIP, ciprofloxacin; CRO, ceftriaxone; PEN, penicillin; SPECT, spectinomycin; TET, tetracycline; (D) Resistome characterisation for each isolate is shown by presence (coloured) or absence (blank) of specified mutations. Dark blue squares indicate gene mutations that have been reported to cause phenotypic resistance, while light blue squares represent mutations of uncertain significance.
{kind=link}
{kind=link}
(A) Median difference in predicted coding DNA sequences (CDS), unfiltered and filtered single nucleotide polymorphisms (SNPs) between any two isolates from the study, two isolates from men within a sexual partnership, and two isolates from separate sites of infection from the one man. Box plots indicate median and IQR, with the whiskers representing the highest and lowest values within 1.5×IQR of the upper and lower quartiles. (B) Median and IQR for pairwise SNP distances between paired isolates from a sexual partnership, and two isolates from the same individual, after filtering for recombination (outlier points not shown).
Only partners in couple 4 were noted to have significantly differing isolates, with a rectal isolate and urethral isolate showing different antimicrobial susceptibility profiles, NG-MAST and MLST results, and differed by over 4000 SNPs, indicating infection with different strains. No other sites were culture-positive for that couple. Two other couples (couples 25 and 26) had isolates that were identical by NG-MAST and MLST, but showed differences at the genome level, with 163 SNPs between partner isolates in couple 25, and 73 SNPs between isolates in couple 26 (online supplementary table S3). After filtering these SNPs for recombination, the differences were less appreciable (1 and 25 SNPs, respectively).
Antibiotic-resistant strains are usually transmitted between partners, rather than arising de novo
To confirm whether antibiotic resistance in N. gonorrhoeae is passed between sexual partners or arises de novo, known genetic determinants of resistance were examined for each isolate. Of couples with the same NG-MAST/MLST strain infecting both partners, isolates from each partner within the partnership had the same phenotypic antibiotic susceptibility, while the corresponding genes or mutations mediating decreased susceptibility or resistance to antibiotics and the adjacent flanking sequences were also identical in the isolate genomes (figure 1, online supplementary table S2). For example, the isolates from both partners in a couple had identical penA alleles, including those with mosaic type alleles associated with penicillin resistance and decreased susceptibility to ceftriaxone. Similarly, where gyrA and parC mutations were present conferring ciprofloxacin resistance in one man, identical mutations were found in his partner’s isolates. The same was true for the plasmid bla TEM-1 beta-lactamase (resulting in penicillinase-producing N. gonorrhoeae) and tetM (resulting in tetracycline resistance) genes.
MSM infected at multiple sites are infected with the same strain at those sites
There were 23 men who had culture-positive N. gonorrhoeae infections at more than one site. In every case (100%; 95% CI 86% to 100%), isolates from different sites within the same man were identical by antimicrobial susceptibility profile, NG-MAST and MLST (figure 1). Using WGS, isolates from the same individual were closely related phylogenetically. However, a small amount of genetic diversity was discernible between isolates within individuals, with up to 22 SNPs differentiating isolates from different sites. Removing predicted recombinant regions excluded the majority of the SNPs distinguishing isolates from the same patient (figure 2).14
Combined genomic analyses provide more detailed inference of gonorrhoea transmission than pairwise SNP distances
Phylogenetic comparisons using WGS data provided higher resolution for determining whether isolates were linked by partner transmission than NG-MAST or MLST. In our data set, there were four instances where multiple couples had isolates with the same NG-MAST and MLST (online supplementary figure S1). In three of these instances, different couples were distinguishable using WGS SNP distances (maximum pairwise SNP distance within couple vs between couples=4 vs 311 SNPs for couples 14 and 17, and 15 vs 36 SNPs for couples 9 and 27, and 2 and 21 SNPs for couples 3 and 6). There were also subtle differences in the patterns of ancestral recombination blocks between isolates from couples 3 and 6 (online supplementary figure S1), suggesting the distribution of SNPs across the genomes differed. In contrast, isolates from couple 14 were unable to be distinguished from the isolates from couple 7, obtained 10 months earlier, using core genome pairwise SNP distances (maximum pairwise SNP distance within couple vs between couples=4 vs 4 SNPs), but had subtly different sequences for the pilus gene pilC2 in the accessory genome. Of the SNPs that differentiated isolates within an individual and from a sexual partnership, approximately half occurred either in non-coding regions or in coding sequences as synonymous mutations. However, of the non-synonymous SNPs in coding regions, the majority of these were in phage-coding or pilin-coding sequences, suggesting these regions are highly variable in the gonococcal genome.
Discussion
In this study, we demonstrated through WGS that MSM transmit their antibiotic-resistant strains of N. gonorrhoeae directly to their partners. The resistance-determining genes/mutations were identical in MSM partners for 33/34 couples, suggesting transmission of the genetic determinants of antibiotic resistance between sexual partners is a key driver behind the spread of antibiotic-resistant gonorrhoea among MSM, who are a major risk group for gonorrhoea. To our knowledge, our study is the first to provide detailed genomic evidence for direct transmission of the genetic determinants for N. gonorrhoeae resistance between men across multiple antibiotic classes at a person-to-person level.
The increasing spread of antimicrobial resistance in N. gonorrhoeae has markedly limited empirical treatment options.1 Genomic epidemiology studies have previously found resistance mutations arising independently in several lineages, with subsequent dissemination.5 6 8 15 16 In each of these studies, highly clonal groups were identified among isolates with resistant phenotypes. More recently, De Silva et al 17 used WGS to track the spread of resistance in N. gonorrhoeae isolates in the UK. However, in contrast to their study and other studies, we examined for direct transmission of multiple resistance determinants between verified sexual partners.
As for other pathogens, WGS has proved a useful tool to understand gonorrhoea transmission. Recently, Didelot et al 18 performed a genomic analysis of two gonorrhoea outbreaks in the UK, while the study by De Silva et al also examined sexual contacts in the UK, mainly from heterosexual networks. These studies revealed some findings parallel to our own observations. In our study, isolates from men within sexual partnerships and isolates from multiple infections within individual men were highly similar based on number of pairwise SNP distance, compared with other isolates in the study. Partner isolates were also more likely to have a similar number of predicted genes (CDS) than two randomly selected isolates from our data set, although the differences in number of CDS between isolates within an individual and between partners were most likely due to assembly/annotation artefact. Our study also showed the genomic diversity among isolates from men within partnerships was also similar to the diversity present within an individual infected at multiple sites. Thus, as shown in heterosexual contacts,17 18 we demonstrated that paired isolates from sexual partners are also usually indistinguishable for MSM, where rectal and pharyngeal infections are more prevalent.19 Due to the absence of symptoms, gonorrhoea at these extragenital sites can remain untreated, potentially providing additional opportunity for mutation by genetic evolution and horizontal exchange of genetic material.1
Our observations that the N. gonorrhoeae strains were indistinguishable between men within sexual partnerships (for 33/34 couples), as well as within each individual man, have important implications for clinical care. First, when men within sexual partnerships are diagnosed with gonorrhoea, clinicians can assume that in most cases the same strain and resistance pattern will be present in both partners. Second, because of this, an antibiotic that is effective in one partner can be empirically used to treat his partner. Third, an antibiotic that is effective at one site of infection is likely to be effective at other sites within the same man, assuming adequate antibiotic penetration. We found little evidence for de novo development of resistance or recent horizontal movement of mobile resistance elements in our cohort, including among high-risk clones previously associated with antimicrobial resistance. Taken together with the findings from previous genomic epidemiology studies of gonorrhoea, our results support the hypothesis that changes in resistance rates follow fluctuations in the prevalence of particular antimicrobial-resistant clones.
There are some caveats to consider when interpreting our study findings. Our observations may not extend to gonorrhoea in heterosexuals, where pharyngeal and rectal infections are generally not as prevalent as for MSM.20 Our study only included the first presentation of the couples during the study period. Given that no history of treatment was recorded, it is possible that there was limited selective pressure applied to promote de novo mutation or acquisition of resistance genes.
Our data also indicate that using the number of SNPs between two isolates may not be the optimal method to verify transmission of gonorrhoea. For example, in our study, two couples had isolates that were indistinguishable by NG-MAST, MLST and pairwise SNP distance. Although it is possible that the couples involved were epidemiologically linked and mixed in the same sexual network, the differing patterns of ancestral recombination blocks and length of time in between sample collection suggest direct transmission between the couples was highly unlikely. In addition, the pairwise SNP distance between isolates from different couples was occasionally less than the maximum pairwise SNP distance between isolates within an individual. Thus, by solely using the number of SNPs between the isolates, we would not have been able to determine definitively whether the isolates from these MSM were transmitted from their partners or from another couple.
The optimal approach to confirm direct transmission of gonorrhoea between individuals with WGS is uncertain. While phylogenetic and Bayesian modelling approaches have been used to compare isolates in outbreak analyses,17 18 these methods require the removal of recombinant regions—sites where mutations have arisen through horizontal acquisition of homologous new genetic material, rather than evolving through stepwise accumulation of nucleotide substitutions—to produce a refined phylogenetic signal.7 This excludes much of the genomic signal available to discriminate isolates, which as shown in our data can be used to differentiate otherwise indistinguishable isolates from different couples. While there is a need to account for horizontal genetic movement, which can introduce large numbers of SNPs with a single mutation event, better approaches to using these data to inform the likelihood of transmission are required.
Another consideration in our study was the selection of single-colony isolates for DNA extraction and WGS, and whether this represented the complete genetic diversity of the underlying microbial population. Through inference from pairwise SNP comparisons of multisite infection and partner isolates (table 2), it is likely that some intrasite and within-host genetic diversity exists. However, these differences appear small, with the exception of true multistrain infection involving different molecular sequence types and antimicrobial susceptibility profiles, which may be missed using single-colony selection. These observations support the findings of De Silva et al 17 who assessed intrasample genetic diversity by selecting and sequencing 12–14 single colonies from six clinical samples selected at random. Further work is required to ascertain whether the subtle genetic variation present in an individual is also reflective of the polyploidy variation present in N. gonorrhoeae.
WGS proved to be a powerful discriminatory tool for distinguishing strains and can be used to provide a high-resolution analysis for identifying transmission networks for gonorrhoea. Use of WGS for gonorrhoea in clinical care has the potential to guide appropriate treatment and public health measures that help limit the further spread of N. gonorrhoeae resistance.
Acknowledgments
The authors would like to acknowledge A Afrizal for his assistance with data extraction, Glenda Fehler for her assistance with samples storage, Samantha Tawil for her assistance with antimicrobial susceptibility testing and Dr Robyn Lee for her knowledge of genetic determinants of resistance in N. gonorrhoeae.
References
Footnotes
MYC and BPH contributed equally.
Handling editor Catherine A Ison
Contributors JCK, EPFC, MYC and BPH designed the study. TS provided analytical tools for genomics analyses. All authors analysed and interpreted the data. JCK primarily drafted the manuscript, with help from MYC and BPH. All authors reviewed and contributed to the final version of the manuscript.
Funding This work was supported by the Victorian Government, Australia, through the Microbiological Diagnostic Unit Public Health Laboratory and the Melbourne Sexual Health Centre. JCK (GNT1074824), EPFC (GNT1091226), TPS (GNT1008549) and BPH (GNT1105905) all received funding from the National Health and Medical Research Council of Australia. JCK is also supported by the NHMRC Centre of Research Excellence in Emerging Infectious Diseases (GNT1102962).
Competing interests MYC is the principal investigator for a gonorrhoea treatment trial funded by Cempra.
Ethics approval Ethics approval was granted by the Alfred Health Human Ethics Committee (approval number 55/15).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Raw sequence data generated for this project have been uploaded to the Sequence Read Archive under the study accession PRJEB17738.