Article Text

Abstract
Background: Virus isolation in cell culture is the recognised diagnostic gold standard for genital herpes. Although increasing evidence indicates that polymerase chain reaction (PCR) provides a more rapid and sensitive diagnostic method, its implementation in routine diagnostic settings has been limited by concerns over contamination and cost.
Objective: To evaluate the feasibility of replacing virus culture with PCR for the diagnosis of genital herpes in settings serving large populations of genitourinary medicine (GUM) attendees.
Methods: Genital swabs collected from 233 consecutive GUM attendees with suspected genital herpes were tested in parallel by virus culture and automated real time PCR. Three specimen preparation methods were evaluated and the assay reliability was assessed by repeat testing, comparison with a commercially available assay, and herpes simplex virus (HSV) sequence analysis. Probe melting temperatures (Tm) were used to differentiate between HSV types without additional post-PCR steps.
Results: HSV was detected in 79/233 (34%) samples by virus culture and 132/233 (57%) samples by PCR. PCR significantly increased HSV detection in both early (<5 days) and late (⩾5 days) presentations and in both first and recurrent episodes. HSV detection and typing by PCR was achieved within less than 4 hours leading to a significant reduction in labour compared to virus culture. Most specimens (120/132, 91%) were typed as HSV-2. Results were highly reproducible.
Conclusions: Real time PCR is a highly reproducible, rapid, and labour efficient method for HSV detection in genital swabs. Its implementation is feasible in routine diagnostic settings.
- BSA, bovine serum albumin
- CPE, cytopathic effect
- FCS, fetal calf serum
- GUM, genitourinary medicine
- HSV, herpes simplex virus
- MEM, minimal essential medium
- PCR, polymerase chain reaction
- PEG, polyethylene glycol
- Tm, melting temperatures
- VTM, viral transport medium
- genital herpes
- polymerase chain reaction
- herpes simplex virus
Statistics from Altmetric.com
- BSA, bovine serum albumin
- CPE, cytopathic effect
- FCS, fetal calf serum
- GUM, genitourinary medicine
- HSV, herpes simplex virus
- MEM, minimal essential medium
- PCR, polymerase chain reaction
- PEG, polyethylene glycol
- Tm, melting temperatures
- VTM, viral transport medium
In 2002, over 18 000 new cases of genital herpes were reported from genitourinary medicine clinics in the United Kingdom, making genital herpes the fourth most common sexually transmitted disease.1 These figures underestimate the true prevalence of symptomatic infections as atypical presentations are common and often misdiagnosed.2 Underdiagnosis of genital herpes is an important obstacle to effective management and control of transmission. The clinical diagnosis of genital herpes is both insensitive and non-specific and requires laboratory confirmation.3,4 Virus isolation in cell culture has long been regarded as the diagnostic gold standard5 and is the diagnostic method of choice in the United Kingdom.6 Virus isolation is slow and labour intensive, requiring up to 7–10 days for the appearance of cytopathic changes in cultured cells or before a negative result can be issued. Specificity is virtually 100%, but levels of virus shedding, quality of specimen, and transport conditions influence sensitivity.7–9 The rate of virus recovery declines significantly with time since the onset of lesions, from 52–93% for vesicles to 41–72% for ulcers and 19–27% for crusted lesions.8,9 Furthermore, lack of specimen refrigeration after collection and during transport markedly reduces virus viability.7
Recent studies have shown that polymerase chain reaction (PCR) increases the rate of HSV detection in mucocutaneous swabs by 11–41% compared to virus culture.9,10 Although earlier PCR techniques were laborious, expensive and prone to contamination, newly developed real time PCR assays are fully automated and allow virus specific detection within a closed system, with fast turn around times and low risk of contamination.9–12 Whereas published protocols for herpes simplex virus (HSV) PCR do not always differentiate between HSV types,10 new technologies allow simultaneous detection and typing of HSV in a single reaction tube, based on software that analyses the different Tm values of HSV-1 and HSV-2 specific probes.11,12 This is a valuable feature for the use of PCR in routine clinical practice, as differentiating between HSV types provides important prognostic information in genital herpes.13–16
The aim of this study was to compare the performance of virus culture and real time PCR for the diagnosis and typing of genital herpes in a clinical setting serving a large population of genitourinary medicine (GUM) clinic attendees. Three different specimen preparation methods were evaluated for their performance, cost, and labour intensity. Sequence variations within the target DNA polymerase gene region were investigated to assess their potential impact on the assay’s performance.
MATERIALS AND METHODS
Patient populations
With ethics committee approval, genital swabs were collected from 233 consecutive GUM clinic attendees who presented with clinical features suggestive of genital herpes at the Caldecot Centre for Sexual Health at Kings College Hospital, London, between June 2002 and August 2002. The first part of the study recruited 145 consecutive patients. Their demographic and clinical data were recorded, including swab site, type of presentation (first or recurrent episode), duration of onset (<5 days or ⩾5 days), presence of ulceration, administration of antiviral drugs before disease onset (aciclovir, valaciclovir, or famciclovir), and HIV status. In the second part of the study, 88 additional genital swabs from consecutive patients presenting with symptoms consistent with genital herpes in the same clinical setting were tested, to provide a validation group for the larger subset.
Sampling
One swab was taken from each patient and placed in 2.5 ml viral transport medium (VTM) containing minimal essential medium (MEM) balanced with 25 mM Hepes (BioWhittaker, Europe) supplemented with 10% bovine serum albumin (BSA), 7.5% sodium bicarbonate, penicillin (500 U/ml), streptomycin (1 mg/ml) and amphotericin B (5 μg/ml). In the laboratory, samples were vortexed for 30 seconds and separated into five aliquots of 500 μl to be used for parallel virus culture and PCR.
Virus culture and typing
For virus isolation, 200 μl of the specimen in VTM were inoculated into two tubes containing a monolayer of Vero monkey kidney cells. Cultures were maintained in MEM-Hepes growth medium (BioWhittaker) supplemented with 25% fetal calf serum (FCS), and incubated at 37°C for 10 days. Cell cultures were examined daily for the appearance of a cytopathic effect (CPE). When a CPE was observed, the cells were scraped off the tube, dried on a slide, fixed in acetone and stained with fluorescein labelled murine monoclonal antibodies against HSV-1 and HSV-2 (Syva Microtak, Palo Alto, CA, USA) according to manufacturer’s instructions.
Specimen preparation for PCR
With a subset of 140 samples, three different methods were used to prepare DNA from 200 μl of specimen in VTM: (1) manual DNA extraction with the QIAamp DNA Mini Kit (Qiagen, UK); (2) automated DNA extraction with the MagNA Pure LC (Roche Diagnostics, Germany), and (3) DNA precipitation with polyethylene glycol (PEG) and sodium chloride (NaCl). The QIAamp DNA Mini Kit was used in accordance with manufacturer’s instructions with the following modifications: an aqueous solution of poly dA (5 μg/ml) (Amersham Biosciences, UK) was added during lysis and 230 μl of 99% ethanol was used at the first wash step to improve extraction in all samples. For the MagNA Pure LC instrument, the sample material was placed into the wells of the sample cartridge, with binding/lysis buffer, proteinase K and extraction carried out according to the manufacturer’s instructions. DNA was eluted in 60 μl of sterile PCR grade deionised water (Sigma, UK) for the QIAamp DNA Mini kit and in 100 μl of elution buffer (Total Nucleic Acid Isolation kit, Roche Diagnostics) for the MagNA Pure LC system. PEG precipitation was performed by adding 200 μl of the specimen in VTM to a mixture of 2.5M NaCl (25 μl) and 20% PEG 8000 (25 μl). The mixture was vortexed, incubated on ice for 30 minutes, and centrifuged at 11 000 g for 20 minutes at 4°C. The supernatant was aspirated, pellet recentrifuged at 4°C for 5 minutes before the pellet was re-suspended in 20 μl of deionised water.
Real time PCR was performed with the LightCycler (Roche Diagnostics) using a method previously described, that amplifies a 215 bp region of the viral DNA polymerase gene.11,12,17 A total volume of 20 μl was obtained by adding 5 μl of DNA or precipitated virus to a PCR reaction mix in glass capillaries. DNA amplification was achieved using the Roche LC Fast Start DNA Master Hybridisation Probe kit (Roche Diagnostics) as described by the manufacturer. Forward (HSV POL F 5′-GCTCGAGTGCGAAAAAACGTTC-3′) and reverse primers (HSV POL A 5′-TGCGGTTGATAAACGCGCAGT-3′) and two fluorescently labelled hybridisation probes HSV-2 FLU (5′-gCgCACCAgATCCACgCCCTTgATgAgC-FAM-3′) and HSV-2 LCR (5′-LC Red 640-CTTgCCCCCgCAgATgACgCC phosphate-3′) (TIB MolBiol, Germany) were used at 5 μM and 1 μM respectively.
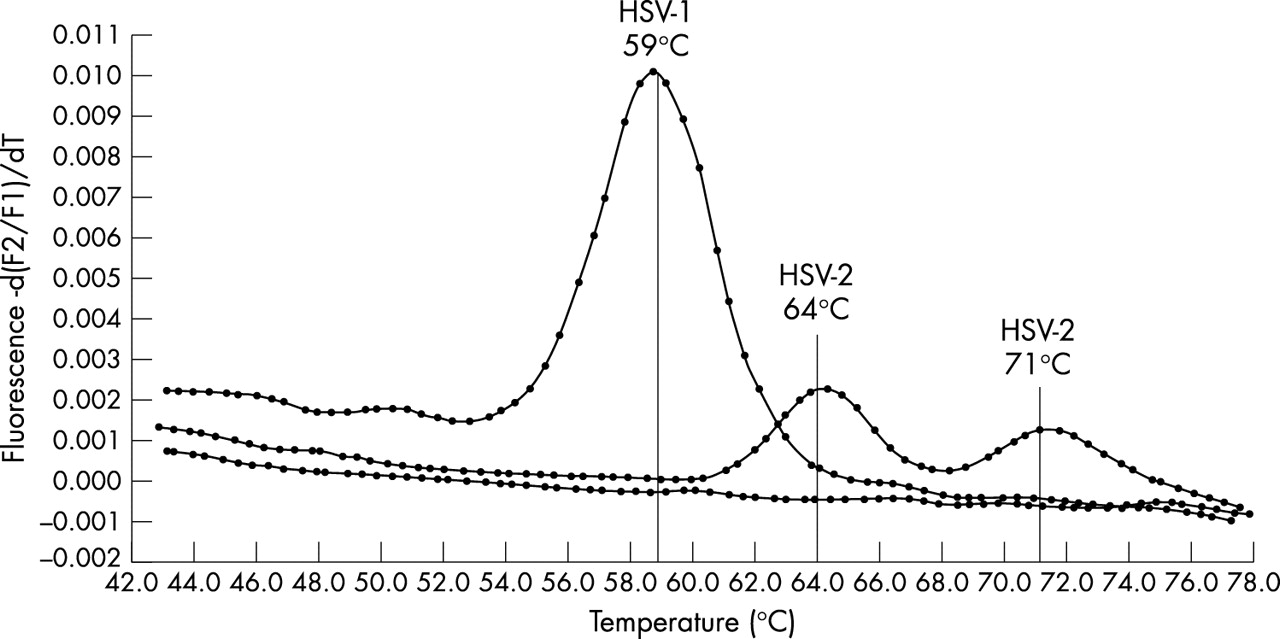
PCR conditions were as follows: initial denaturation at 95°C for 10 minutes (temperature transition rate of 20°C/s) to activate DNA Taq polymerase, followed by 50 cycles of denaturation at 95°C for 5 seconds (20°C/s transition), annealing at 50°C for 5 seconds (20°C/s transition) with single fluorescence acquisition, and extension at 72°C for 5 seconds (20°C/s transition). Melting curve analysis was performed as follows: annealing at 40°C for 30 seconds (20°C/s transition) followed by an increase to 80°C for 0 second (0.1°C/s transition) with continuous fluorescence acquisition, and cooling to 40°C for 30 seconds (20°C/s transition). The Tm of DNA fragments and hybridisation probes was used to differentiate between HSV types. The probe Tm was 59°C for HSV-1 and 64°C and/or 71°C for HSV-2. The RealArt HSV1/2 LC PCR kit (Artus Biotech, Germany) was used according to the manufacturer’s instructions to confirm the results of 15 HSV positive specimens.
DNA sequencing
Samples were prepared for LightCycler PCR as described earlier but in the absence of hybridisation probes HSV-2 FLU and LCR. The final volume of 20 μl was made up with deionised water. Following amplification, the DNA was purified using QIAquick PCR Purification kits (Qiagen) according to manufacturer’s instructions and eluted in 50 μl of deionised water.
DNA sequencing was performed by the Sanger dideoxy chain termination method using 3 μl of extracted DNA, 1 μl of each A, C, G, T reagent mix from the ThermoSequenase DYEnamic Direct Cycle sequencing kit (Amersham Pharmacia Biotech Inc, UK), 1 μl of 0.5 μM HSV forward and reverse primers (as previously described) labelled with CY5.5 and CY5 respectively (Sigma-Genosys Ltd, UK). Reactions were performed on a GeneAmp 9700 thermocycler (PerkinElmer Applied Biosystems, Beaconsfield, UK) under the following conditions: 25 cycles of denaturation at 94°C for 5 minutes, followed by further denaturation at 94°C for 30 seconds, annealing at 50°C for 30 seconds, and extension at 70°C for 1 minute. Formamide loading dye (7 μl) was added, and the DNA denatured at 80°C for 2 minutes. Automated DNA sequencing reactions (2 μl) were run on an Trugene HIV-1 Opengene sequencing system (Visible Genetics, Ontario, Canada) under standard conditions using version 3.16 software as described by the manufacturer. Sequence data were compared to other HSV sequences present in the GenBank database to confirm HSV type using BLAST.18 Sequences were aligned using ClustalW version 1.81.19
Analysis of results
The HSV detection rates for virus culture versus PCR according to clinical characteristics were compared by the χ2 or Fisher’s exact test as appropriate. In the comparison of specimen preparation methods, the “true positive” reference was specimens that were either positive in both virus culture and PCR, or were virus culture negative but PCR positive in ⩾2 separate assays and by ⩾2 methods. Kappa values were calculated to determine the level of agreement of the three specimen preparation methods when compared to the reference.
RESULTS
In the first part of the study genital swabs were collected from 145 consecutive patients (85 men and 60 women) with suspected genital herpes, and tested in parallel by virus culture and PCR. The most common swab sites were the penile skin in males (63/85, 74%) and the vulva in females (48/60, 80%). HSV was detected in 45/145 (31%) specimens by culture and 77/145 (53%) specimens by PCR. The 32 specimens that were negative by culture but positive by PCR underwent extensive retesting. They were all confirmed HSV DNA positive in ⩾2 independent replicate PCR assays (mean 2.75 assays per sample). In addition, with a random subset of 15 samples, the in-house PCR results were confirmed by a commercially available PCR assay. Thus real time PCR increased the HSV detection rate by 71% (32/45). High reproducibility indicated a low risk of false positive results. No patient with positive culture had a negative PCR. The average time for a positive virus culture was 3 days (range 2–10 days), whereas negative results required 10 days of incubation. PCR results required less than 4 hours including time for specimen preparation, amplification, detection, analysis, and typing.
Table 1 shows the results of virus culture and PCR in relation to clinical presentation. By either method the rates of HSV detection were similar in men and women, and PCR increased HSV detection by 79% (19/24) and 62% (13/21) in the two groups respectively. Culture was more frequently positive in patients presenting <5 days of onset than in those who presented later (27/64, 42% versus 18/81 22%, p = 0.01). The proportion of patients with a positive PCR did not differ significantly between the two groups (38/64, 59% versus 39/81, 48%, p = 0.2). As a result, PCR increased HSV detection by 41% (11/27) in early disease and 116% (21/18) in late disease. The proportion of patients with a positive culture was similar in first episode and recurrent disease. PCR increased HSV detection by 58% (15/26) and 89% (17/19) in the two groups respectively. Improved detection by PCR was seen regardless of HIV status.
Comparison of virus culture (VC) and PCR for HSV detection in different clinical presentations of genital herpes
We subsequently extended the study to compare virus culture with PCR in further 88 consecutive genital swabs obtained from symptomatic patients in the same clinical setting. Of these, 34/88 (39%) were positive by culture and 55/88 (63%) by PCR. Thus, in this additional subset of samples, PCR increased HSV detection by 62% (21/34), validating the data from the larger subset. In the whole cohort, HSV detection rate was 79/233 (34%) by virus culture and 132/233 (57%) by PCR.
HSV typing by LightCycler PCR
Figure 1 shows the reference Tm for HSV-1 and HSV-2. Among the first set of HSV positive samples, 71/77 (92%) were typed as HSV-2 and 6/77 (8%) as HSV-1. All typing results were confirmed in the replicate experiments. The 15 samples tested by the commercial PCR assay also yielded typing results identical to those obtained by the in-house assay. Among the second set of HSV positive samples, 49/55 (89%) were typed as HSV-2 and 6/55 (11%) as HSV-1. In the whole cohort, 120/132 (91%) HSV positive samples were typed as HSV-2. Fourteen randomly selected HSV-2 positive samples with Tm >60°C were sequenced and all confirmed as HSV-2 after alignment to known HSV-2 sequences (table 2). Among the 14 samples sequenced, four showed sequence variation in the FLU probe binding site within the 100 bp region of the DNA polymerase gene (table 2). No variation was observed in the target HSV-2 sequence among the 10 remaining specimens.
Nucleotide sequences of HSV positive specimens with Tm >60°C. Probes are boxed and nucleotide differences are bold

{kind=link}
LightCycler melting curve analysis for HSV typing.
Feasibility of PCR testing for large volumes of samples
As specimen preparation is the most time consuming step in PCR testing, we compared the performance, labour intensity, and cost of three specimen preparation methods using manual DNA extraction with the QIAamp DNA mini kit, automated DNA extraction with MagNA Pure LC, and virus precipitation with PEG. In a comparison of 140 specimens, 120/140 (86%) were either negative or positive by all three methods, 12/140 (8%) were positive by two methods, and 8/140 (6%) were positive by only one method. Of the latter eight samples, all were confirmed positive by repeated specimen preparation and PCR testing, three were also positive by virus culture. Regarding as “true positive” specimens that were either positive by both virus culture and PCR or, if virus culture negative, positive in ⩾2 separate PCR assays and by ⩾2 methods, the HSV detection rate was PEG precipitation >MagNA Pure LC >QIAamp DNA mini kit (table 3). Kappa values for the three specimen preparation methods were ⩾0.84, indicating a high level of agreement. PEG precipitation detected 65/72 (90%) positive samples, including three samples positive by virus culture but negative by the other two preparation methods, and was more rapid and less expensive than the other two methods (table 3).
Comparison of detection rate, duration and cost of three PCR testing methods and virus culture
DISCUSSION
A virological confirmation of genital herpes provides the basis for appropriate management, and informs prognosis and counselling. To date, the routine diagnosis of genital herpes has mainly relied on virus isolation in cell culture. However, numerous recent reports have highlighted the limitations of this method.9 On practical consideration, virus culture is time consuming and requires highly stringent conditions for specimen transport and storage to preserve virus viability.7 Improved diagnostic methods are needed to improve recognition of genital herpes. In this study we demonstrated that real time PCR significantly increased the rate of HSV detection in genital swabs compared to virus culture and that its implementation is feasible for routine diagnostic settings.
PCR was shown to increase the overall rate of HSV detection by 61–71%. Even in patients presenting with visible genital ulcerations PCR detected 88% more infections than virus culture. Thus, in the absence of PCR testing, 53 of the 233 symptomatic patients would have received a negative result, preventing appropriate counselling on prognosis and risk of transmission. Our result is striking considering that a similar study by Scoular9 showed a 24% increased detection rate by PCR over virus culture. This observation may in part be because our study was conducted during summer months. Warm weather may have affected HSV viability after specimen collection and further diminished the sensitivity of virus culture.7
The increase in detection afforded by PCR was observed in different clinical presentations including both first and recurrent episodes. In contrast with previous observations,20 we did not observe a different yield of virus culture in patients with first episode compared with those with recurrent disease. Predictably, the increase in detection by PCR was especially high among patients presenting late (⩾5 days) after the onset of symptoms, as declining levels of virus shedding in older lesions would favour the more sensitive technique.9,20 None the less, detection also increased significantly in patients who presented within 5 days of onset. The absence of a significant difference between culture and PCR yields in patients without visible ulceration is interesting, but the small number of observations precludes speculations about its significance.
Unlike a previous study by Scoular et al,10 the PCR method we used offered the advantage of providing HSV typing without additional, time consuming post-PCR steps. Sequencing of a subset of HSV-2 positive samples confirmed the accuracy of typing. The low level of sequence variation observed indicated high reliability of the assay. Among 132 HSV positive specimens, 91% were HSV-2 and 9% were HSV-1. HSV-2 accounted for the majority of both first and recurrent episodes. These findings are in contrast with results from other centres in the United Kingdom, which report HSV-1 to be responsible for more than ⩾50% of newly diagnosed cases of genital herpes.21–23
To investigate the feasibility of implementing HSV PCR for large volume testing, three specimen preparation methods were compared to evaluate performance, labour, and cost. A high level of agreement was observed between the three methods. Virus precipitation by PEG, although not previously used for LightCycler PCR, had the highest detection rates, was the least expensive, and required the shortest incubation period, thus providing a valid option for specimen processing before PCR. Even though PCR has demonstrated increased sensitivity over virus culture, its implementation into routine clinical practice has been limited by concerns of contamination. Extensive retesting of positive samples in our study demonstrated that real time PCR assays are reliable and highly reproducible. This is a result of amplification occurring within a closed system, with a minimal risk of carryover contamination, although reproducibility was also helped by the high prevalence of disease in our sample.24 Although the costs of consumables for PCR (£2.44–£9.27) are higher than those for virus culture (£1.00–£1.55), real time PCR proved cost efficient by significantly reducing labour cost per sample.
With high levels of asymptomatic shedding and high prevalence of non-classic presentations, laboratory investigations provide guidance for patient management and controlling the spread of HSV infection. The sensitivity of nucleic acid amplification tests exceeds conventional laboratory methods to detect HSV, thus enabling greater diagnostic accuracy. Although virus culture continues to be the standard diagnostic test used in routine practice in the United Kingdom (D Brown and AM Geretti, unpublished observation), its ability to detect HSV in a large proportion of infected individuals is clearly deficient compared to PCR. Our data strongly support the implementation of PCR for the routine diagnosis of genital herpes.
Key messages
-
Real time PCR is a highly reproducible, rapid, and labour efficient method for HSV detection in genital swabs
-
The ability of virus culture to detect HSV in a large proportion of infected individuals is clearly deficient compared to PCR
-
The implementation of real time PCR for the detection of HSV in genital swabs is feasible for routine diagnostic settings
Acknowledgments
The authors thank Dr Benedict Murdin (University of Surrey in Guildford, UK), for statistical support.
CONTRIBUTORS Study design: AMG, MS, MT-F; experimental work: MR, DT, SMl, MS; coordination of sampling and clinical data collection: CMcD; analysis of results: MR, AMG, MS; preparation of manuscript: MR, AMG.
REFERENCES
Footnotes
-
Conflict of interest: None declared.
Linked Articles
- Brief Encounters